
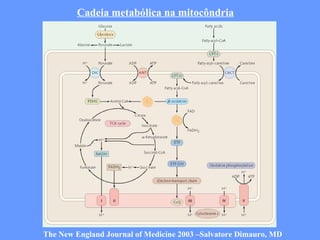





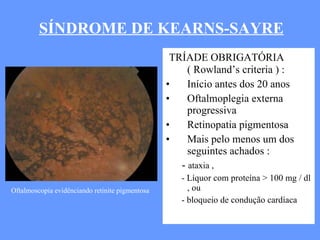
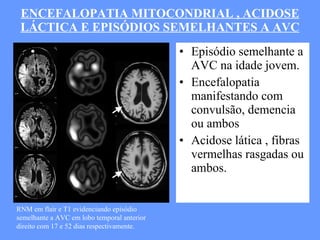

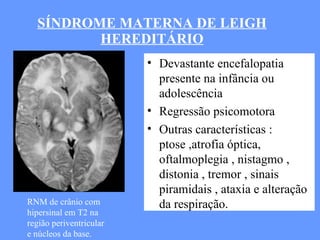
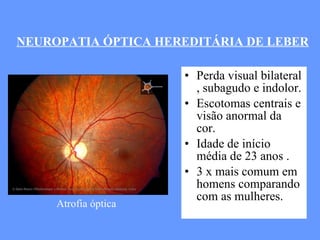

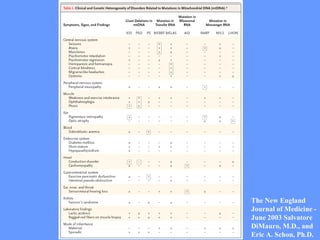






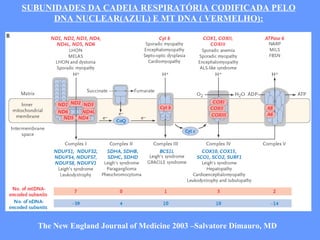

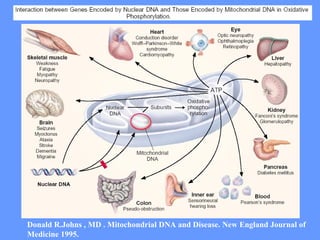


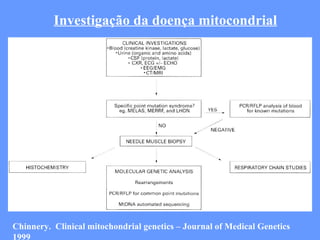



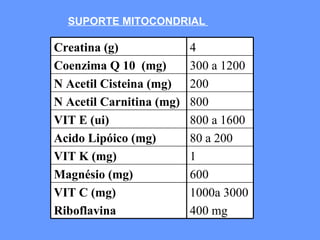
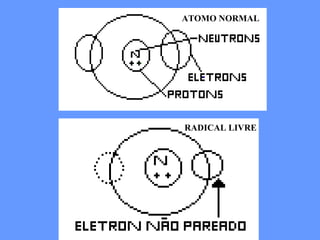
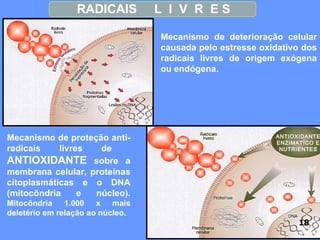
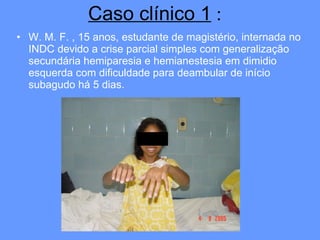
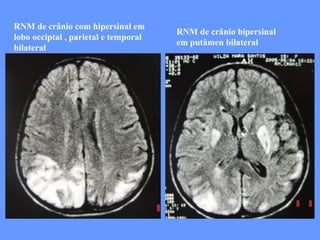


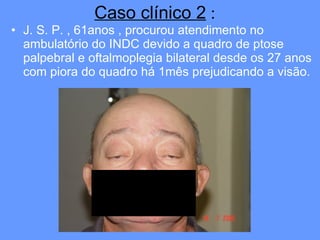


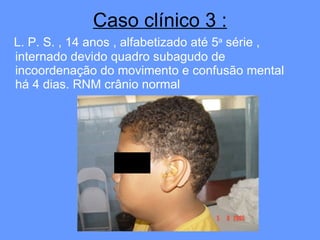



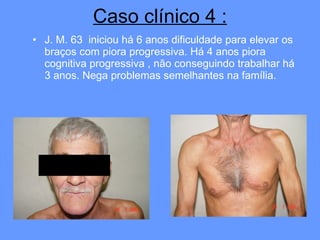

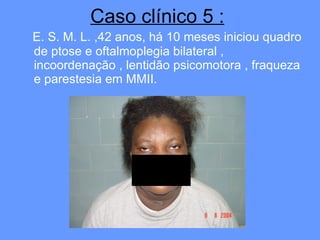




O documento aborda as doenças mitocondriais, incluindo epidemiologia, características clínicas, testes diagnósticos e opções de tratamento. As doenças não são tão raras quanto se pensava, com prevalências que variam entre 10 a 15 casos por 100.000 pessoas, e incluem síndromes como MELAS e MERRF. O tratamento pode envolver terapia sintomática, terapias medicamentosas que potencializam a função mitocondrial e terapia gênica.































































































