Baixado 47 vezes



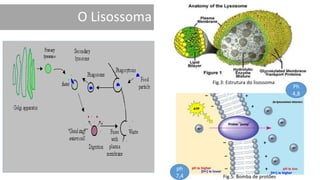
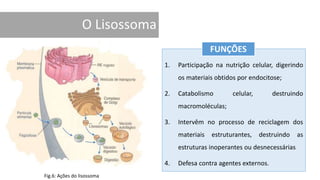

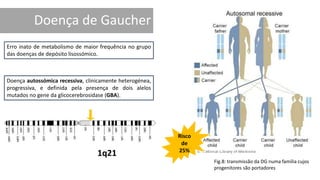
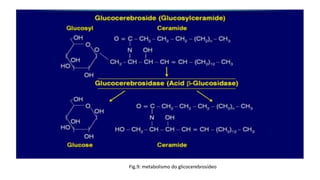
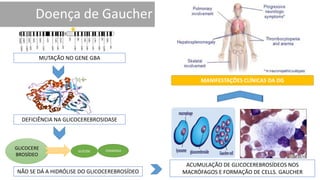
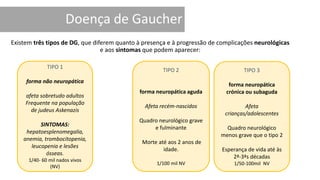

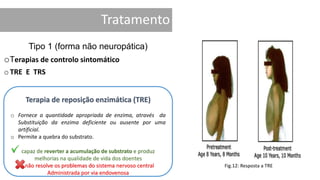
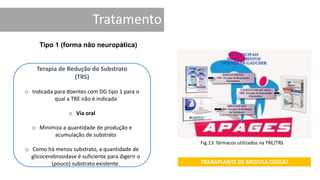
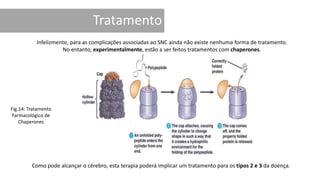
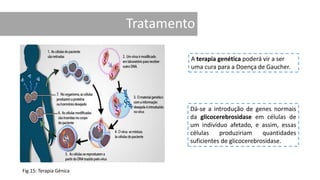




1. A doença de Gaucher é causada pela deficiência da enzima glicocerebrosidase, levando à acumulação de glicocerebrosídeos nos lisossomos. 2. Existem três tipos clínicos da doença dependendo da presença de complicações neurológicas. 3. O tratamento inclui terapia de reposição enzimática e de redução do substrato para o tipo 1, enquanto tipos 2 e 3 ainda não têm tratamento eficaz para complicações neurológicas.

































































