Transferir como PDF, PPTX





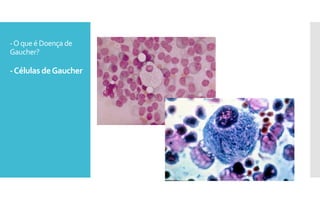
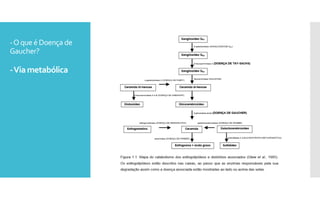
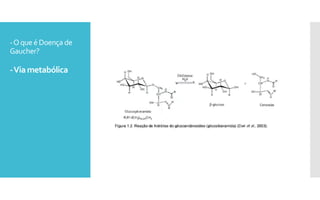


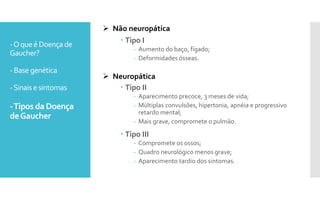
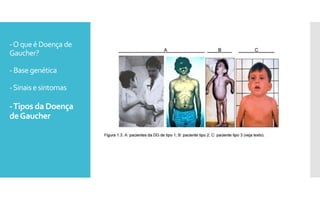










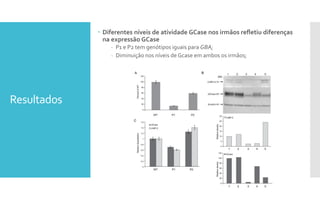
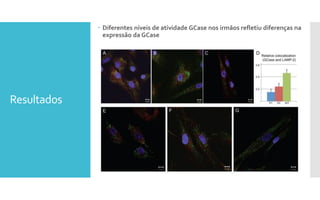

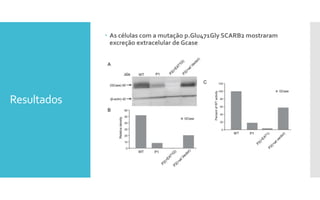






O documento descreve como uma mutação no gene SCARB2, que codifica a proteína LIMP-2, modifica os sintomas da doença de Gaucher em dois irmãos que compartilham as mesmas mutações no gene GBA. A mutação SCARB2 foi encontrada apenas no irmão que apresentava epilepsia mioclônica grave, sugerindo que a LIMP-2 é um modificador da doença de Gaucher.





















































