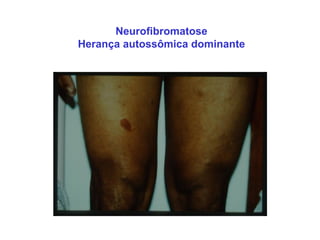
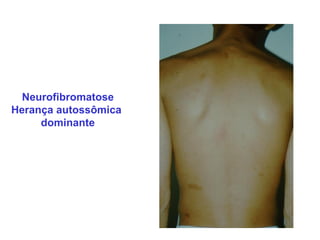
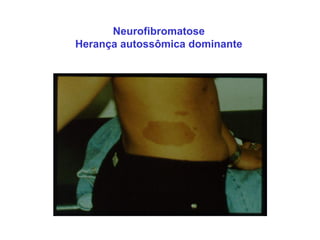
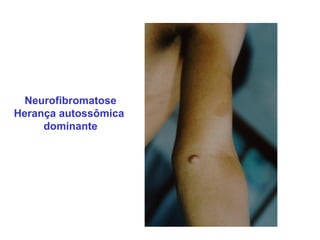
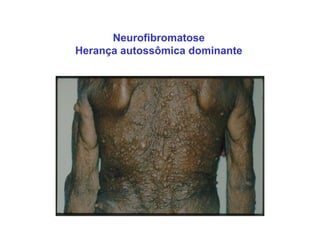
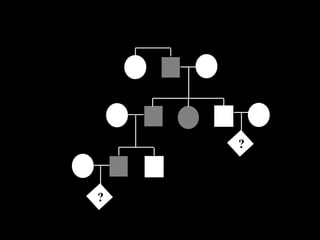
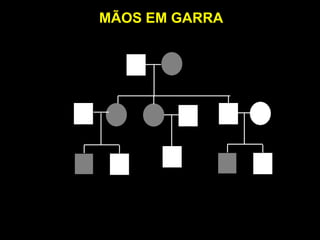
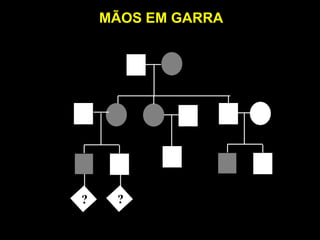
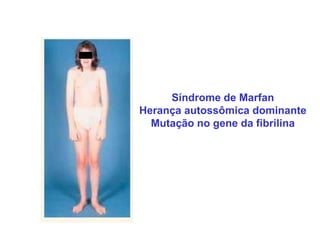
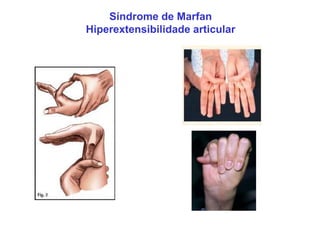
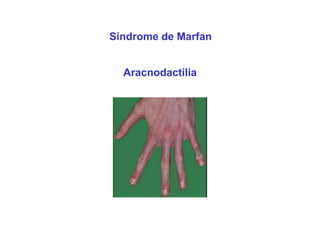
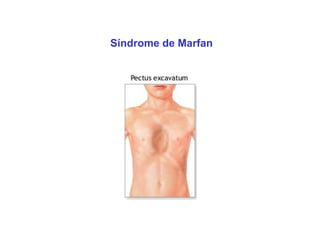
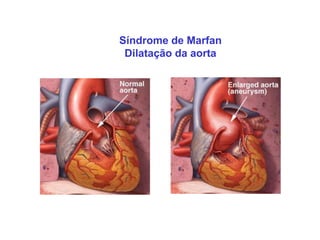
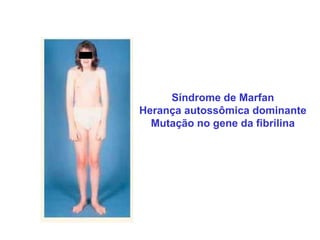
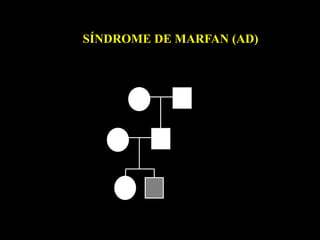
ACONDROPLASIA
CARACTERÍSTICAS CLÍNICAS:
NANISMO DE MEMBROS CURTOS
COM FÁCIES TÍPICA
INCIDÊNCIA: 1 / 15.000
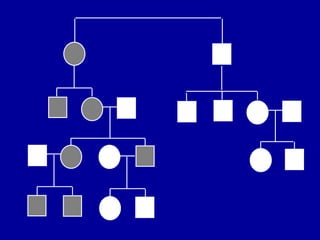
GENÉTICA:
AUTOSSÔMICA DOMINANTE
> 90% DOS CASOS SÃO
ESPORÁDICOS
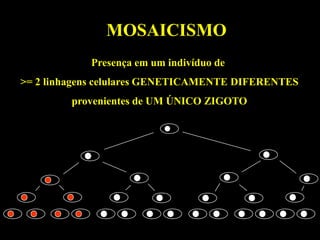
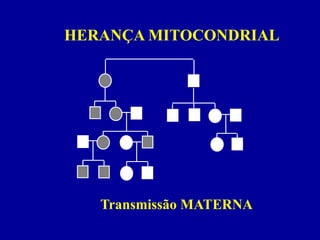
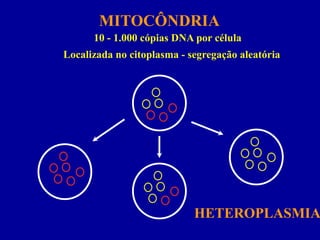
MITOCÔNDRIA
Gera energia celular(ATP) - fosforilação oxidativa
Localizada no citoplasma - segregação aleatória
Contém DNA próprio
32.
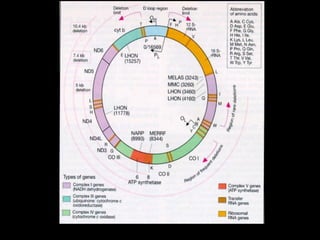
DNA MITOCONDRIAL
DNA NÃO NUCLEAR !
• circular - 16.5 kb
genes de tRNA, rRNA, enzimas fosforil. oxid.
• 5 - 10 cópias / mitocôndria
• 2 - 100 mitocôndrias / célula
• Taxa de mutação 20 x maior
34.
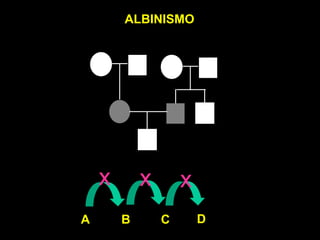
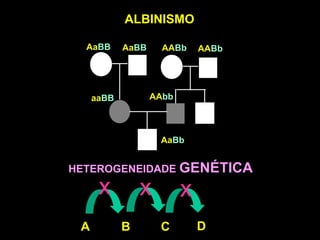
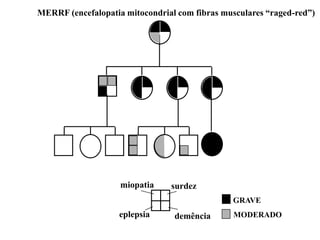
DOENÇAS MITOCONDRIAIS
Energia celular (ATP) - fosforilação oxidativa
• Tecidos mais suscetíveis: coração
músculo esquelético
sistema nervoso central
MIOPATIAS e ENCEFALOPATIAS
• Início tardio e PROGRESSIVAS
FATOR LIGADO À IDADE
35.
DOENÇAS MITOCONDRIAIS
VARIABILIDADE DE QUADRO CLÍNICO
NARP Síndrome de Leigh
fraqueza muscular, ataxia, INÍCIO PRECOCE
retinitus pigmentosa ataxia, hipotonia,
deselvolv. retardado,
atrofia óptica,
degeneração ganglia basal
LETAL
MESMA MUTAÇÃO !! (ATP6 - ATP synthase)
36.
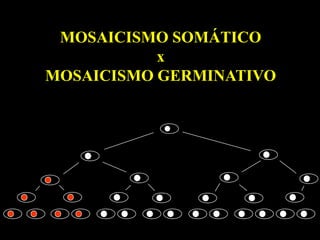
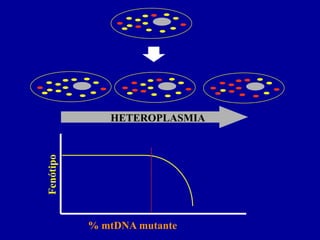
MITOCÔNDRIA
10 - 1.000 cópias DNA por célula
Localizada no citoplasma - segregação aleatória
HETEROPLASMIA
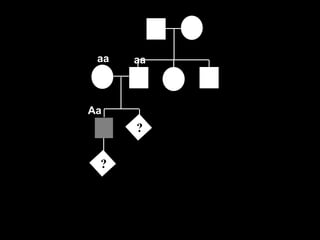
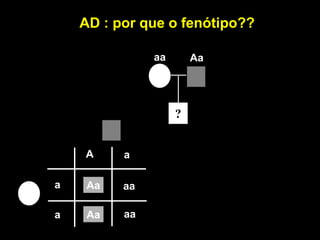
AD : porque o fenótipo??
aa Aa
?
A a
a Aa aa
a Aa aa
42.
HIPERCOLESTEROLEMIA FAMILIAR -DOSAGEM
CARACTERÍSTICAS CLÍNICAS:
HETEROZIGOTOS: nível elevado de
lipoproteína de baixa densidade (LDL) no
plasma, depósito de colesterol nos
tendões e pele (xantomas) e artérias na
idade adulta, doença das artérias
coronárias no início da meia-idade.
HOMOZIGOTOS: manifestações clínicas
mais cedo, doença das artérias coronárias
geralmente fatal na infância.
IMPORTÂNCIA:
uma das doenças genéticas mais comuns
e importante causa de doenças das
artérias coronárias.
INCIDÊNCIA:
HETEROZIGOTO: 1/500
HOMOZIGOTO: 1/1.000.000
GENÉTICA:
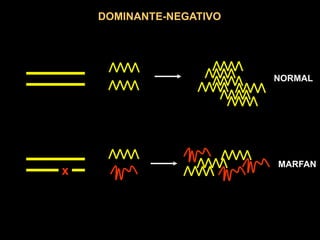
AUTOSSÔMICA DOMINANTE
MÚLTIPLOS ALELOS MUTANTES
MUTAÇÕES NO GENE QUE
CODIFICA O RECEPTOR DA LDL