Baixado 41 vezes









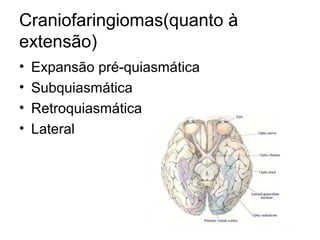




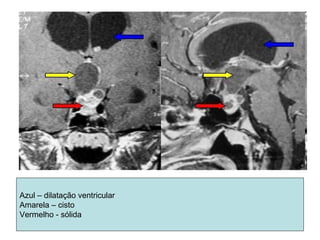




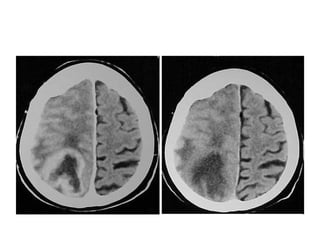
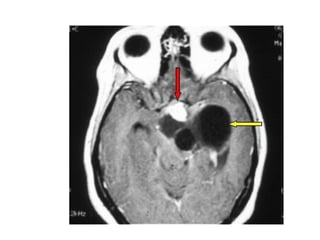











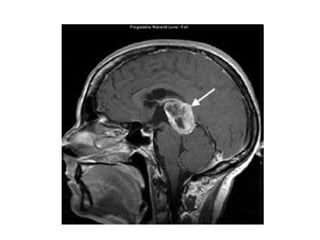













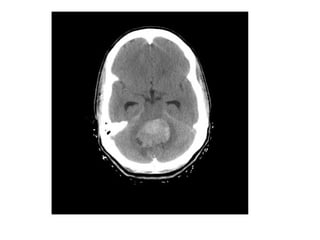








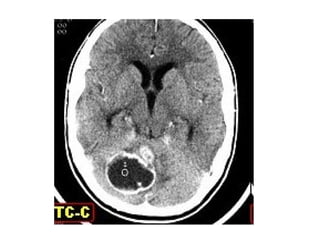





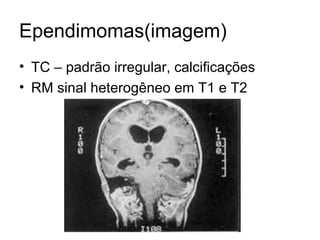




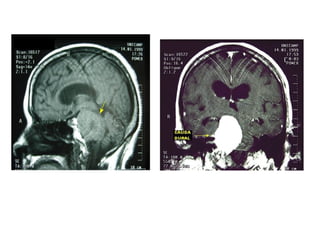



O documento descreve vários tipos de tumores intracranianos em crianças, incluindo seus sintomas, achados de imagem e tratamento. Resume três tumores específicos: 1) Craniofaringiomas são tumores benignos localizados na região suprasselar que causam dor de cabeça e alterações endócrinas; 2) Meduloblastomas são tumores malignos localizados na fossa posterior que causam ataxia e hidrocefalia; 3) Astrocitomas cerebelares são tumores localizados no cerebelo.





















































































