Baixado 26 vezes









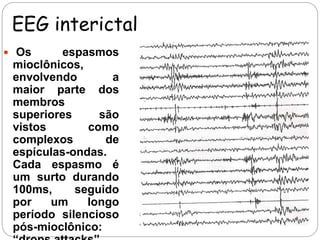







O documento descreve a Síndrome de Doose, uma epilepsia caracterizada por mioclonias que causam quedas. Ela geralmente começa entre 1-5 anos e é causada por fatores genéticos. Os principais sintomas incluem mioclonias, crises tônico-clônicas e de ausência. O tratamento de primeira linha envolve valproato, podendo usar lamotrigina ou etosuximida.



































































