Baixado 125 vezes










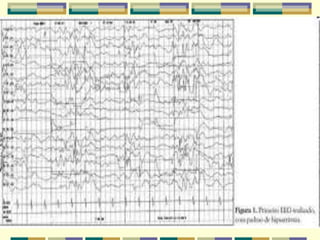

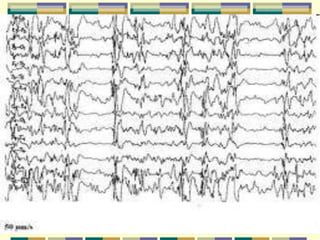























A síndrome de West é caracterizada por espasmos epilépticos em grupos associados a um EEG hipsarrítmico. A síndrome de Ohtahara se inicia no primeiro mês de vida com crises tônicas breves e evolui para padrão de surto-supressão no EEG, podendo evoluir para síndrome de West. O documento descreve essas síndromes epilépticas infantis e seu diagnóstico e tratamento.






































![Exame neuro infantil[1].2](https://cdn.slidesharecdn.com/ss_thumbnails/exameneuroinfantil1-2-120322123912-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)
















































