Baixado 225 vezes



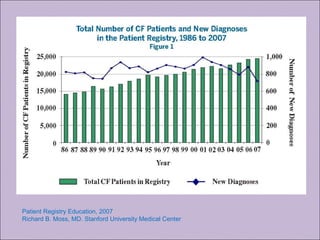

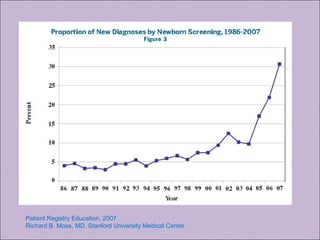
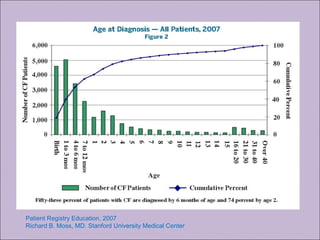
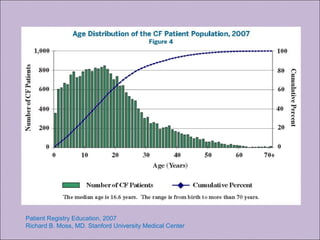
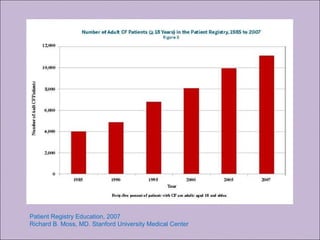
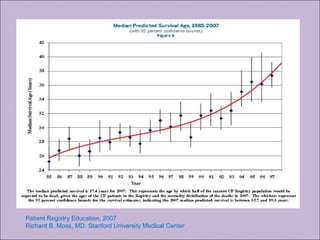

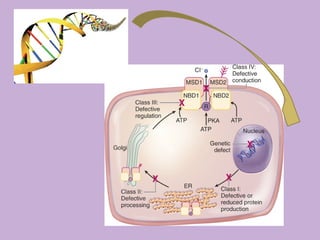
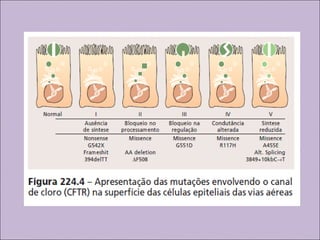
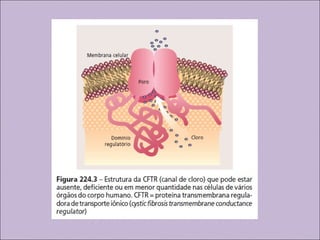
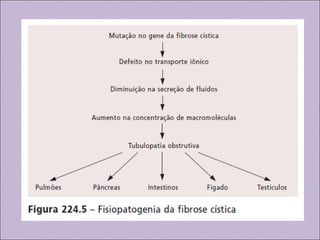
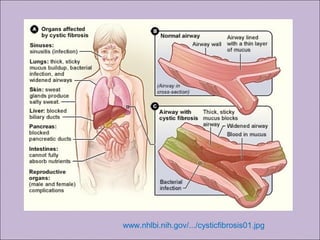


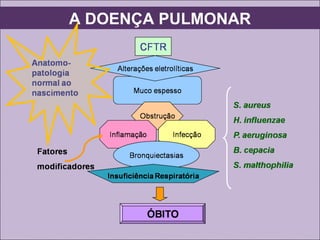
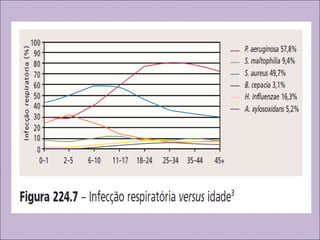


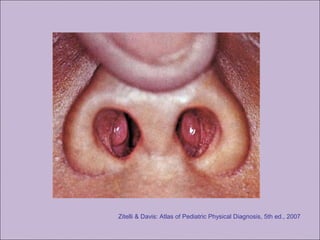
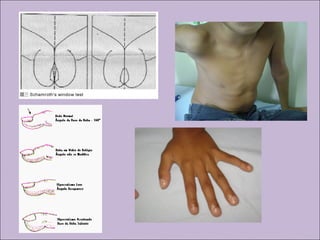
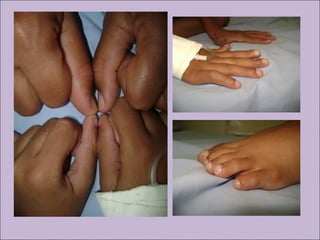
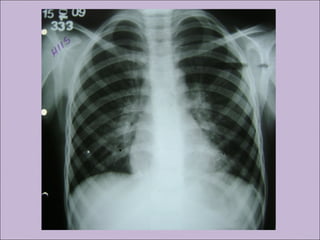
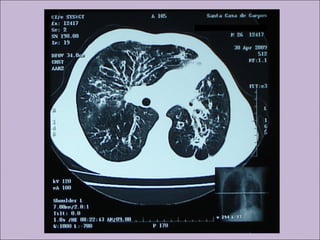
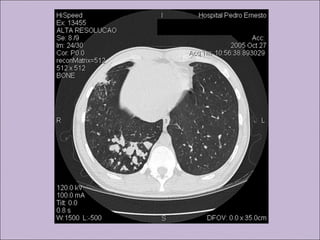
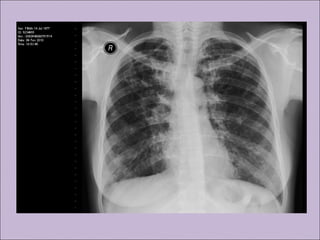
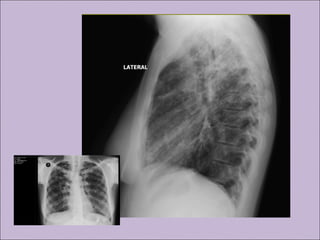
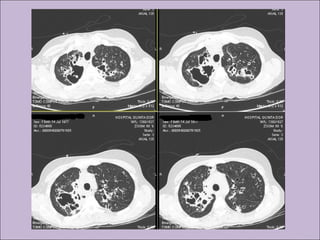
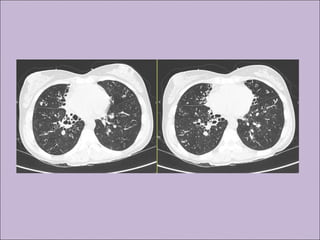
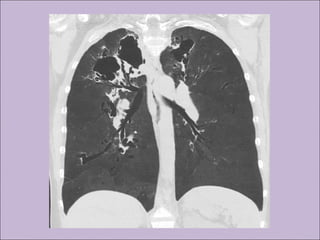
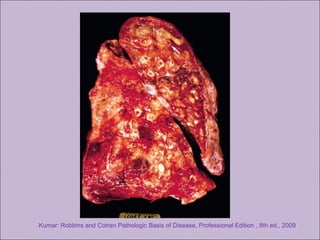

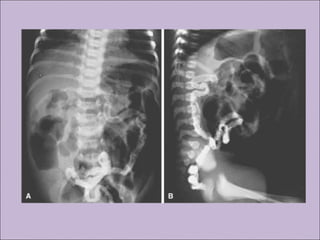
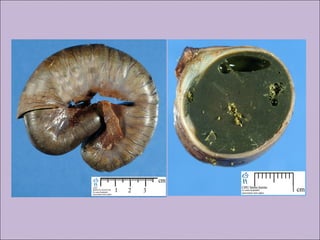

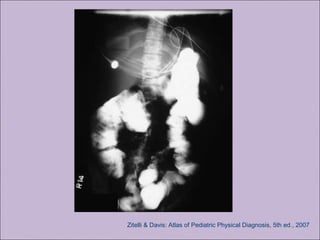





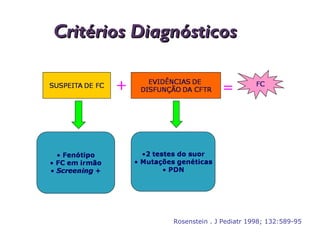
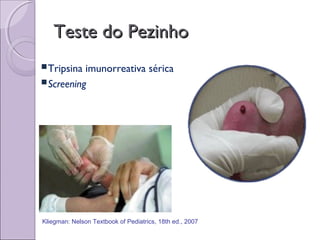

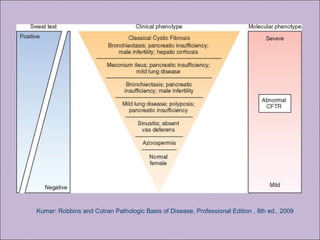






Este documento fornece informações sobre a fibrose cística, incluindo sua epidemiologia, manifestações clínicas, critérios diagnósticos e princípios de tratamento. A fibrose cística é uma doença genética que afeta principalmente os pulmões e sistema digestivo, causada por mutações no gene CFTR. Os sintomas variam de acordo com a faixa etária e incluem problemas respiratórios e gastrointestinais. O diagnóstico é feito através de testes como o do suor e genético.















































































