Baixado 748 vezes






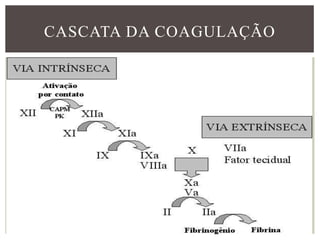
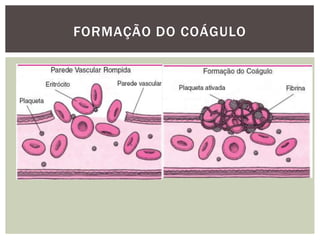


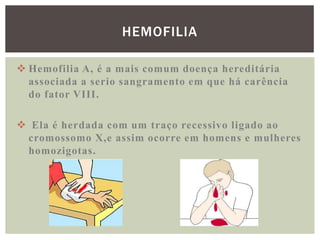















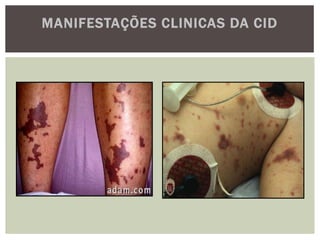






























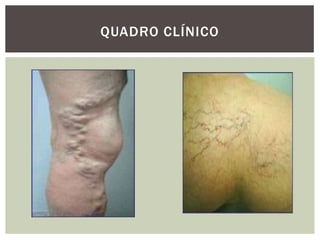





O documento discute distúrbios da coagulação do sangue como a hemofilia, coagulação intravascular disseminada e seus sintomas. A hemofilia ocorre quando faltam os fatores VIII ou IX da coagulação, levando a sangramentos espontâneos ou após trauma. A coagulação intravascular disseminada envolve a ativação difusa da coagulação dentro dos vasos, podendo ser causada por traumas, infecções ou câncer.






![[c7s] Hemofilia](https://cdn.slidesharecdn.com/ss_thumbnails/hemofilia-111129143931-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)





































































