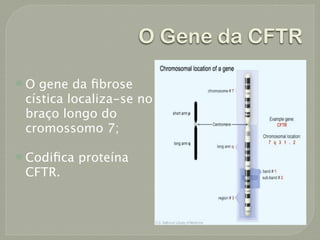
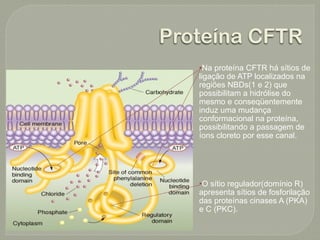
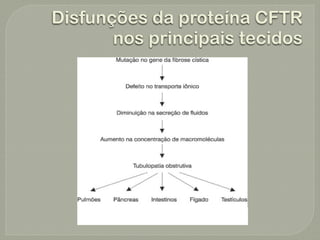
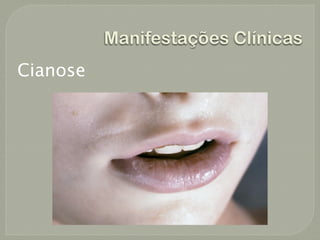
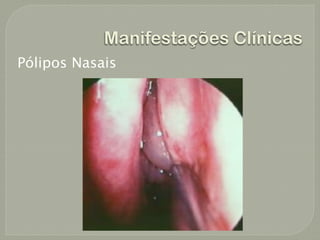
O documento discute a fibrose cística, incluindo sua incidência, patogênese, classes de mutações no gene CFTR, manifestações clínicas, diagnóstico e tratamento. A fibrose cística é uma doença genética rara que afeta principalmente os pulmões e pâncreas, causando secreções espessas que podem levar a infecções e danos progressivos aos órgãos. O diagnóstico é baseado em critérios clínicos e níveis elevados de sal no suor. O tratamento envolve f