Baixado 26 vezes
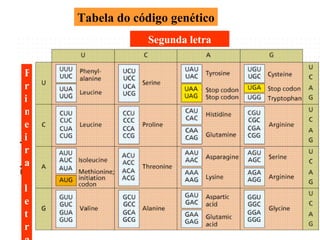
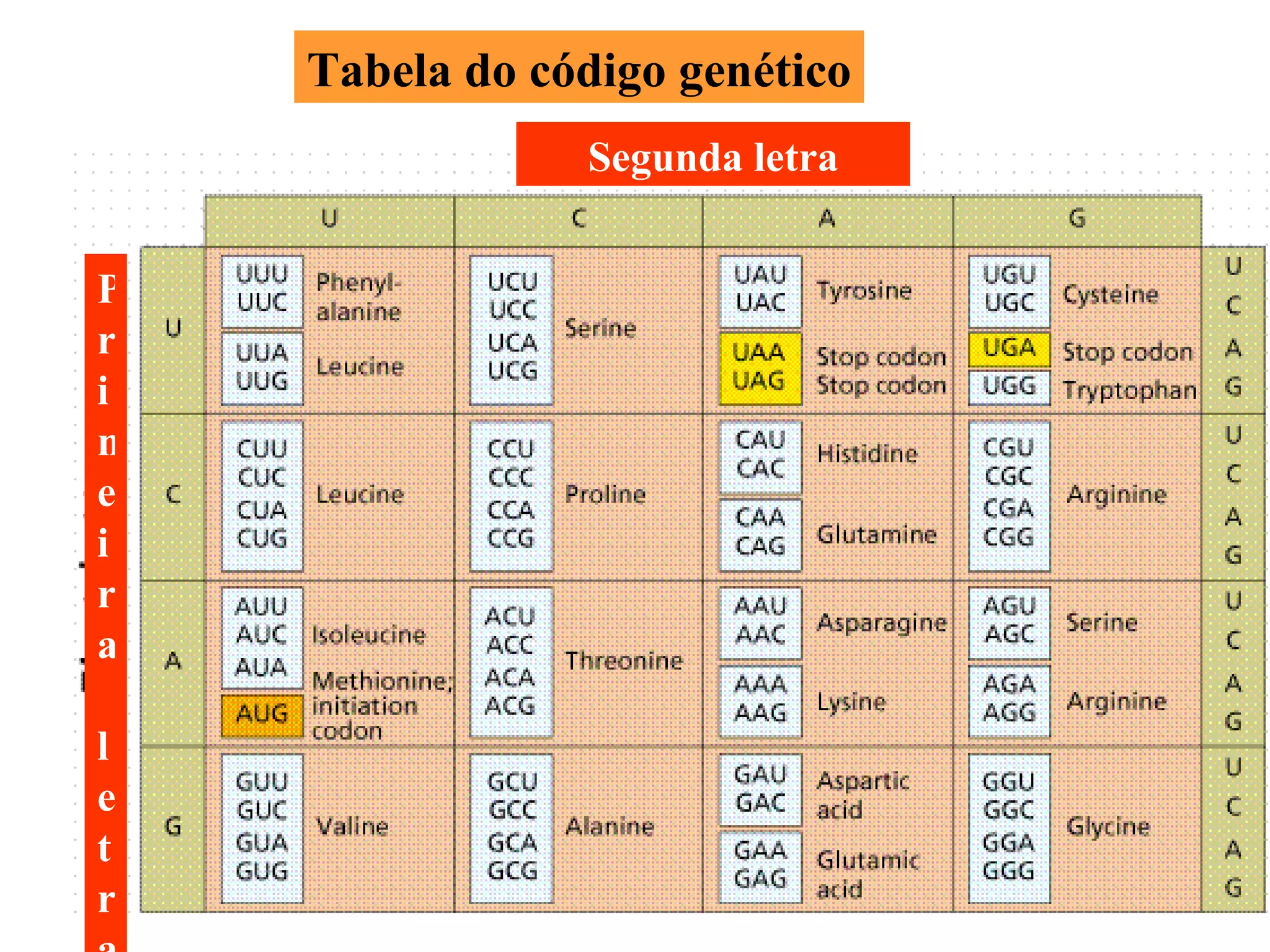

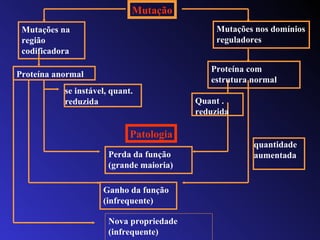
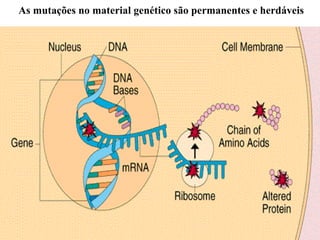
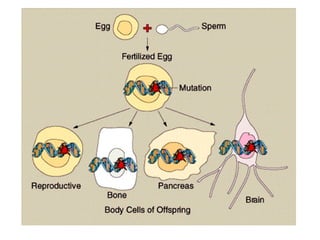

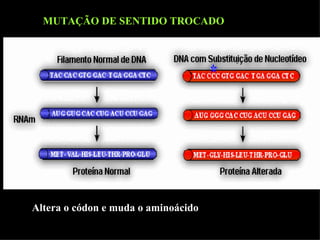

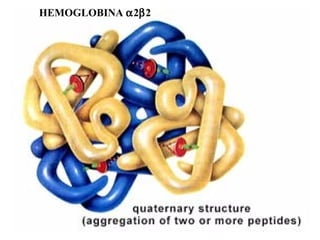
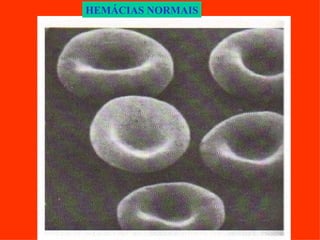

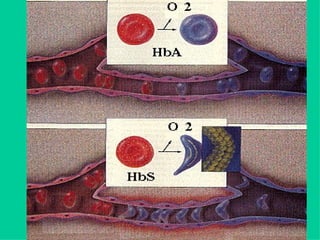
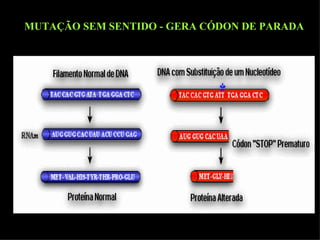

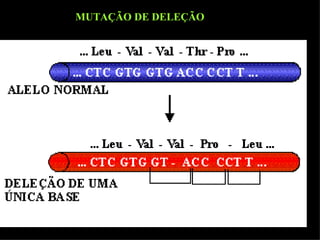
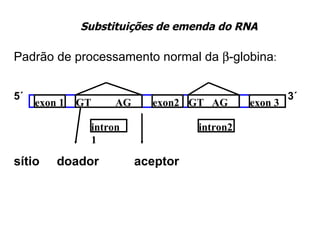

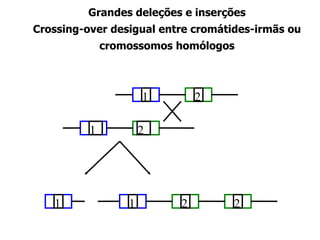

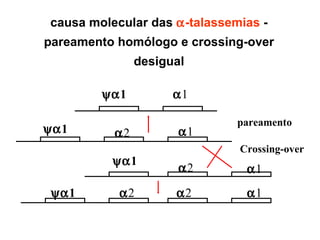
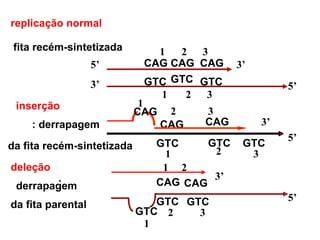
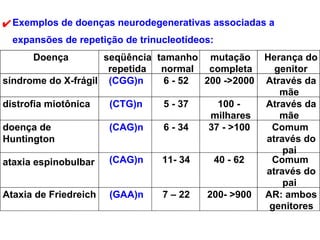
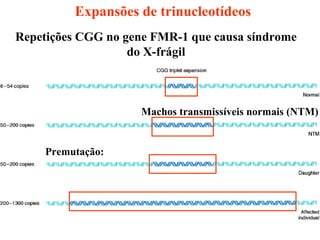

O documento discute os tipos e causas de mutações genéticas, incluindo erros na replicação do DNA, exposição a agentes mutagênicos, substituições de bases, inserções, deleções e expansões de repetições de trinucleotídeos, e como essas mutações podem levar a alterações na estrutura e função de proteínas causando doenças como a doença de Tay-Sachs e a distrofia miotônica.








































![Aula 4 - 41528 Patologia Molecular[1].ppt](https://cdn.slidesharecdn.com/ss_thumbnails/aula4-patologiamolecular1-250607044617-8497f166-thumbnail.jpg?width=640&height=640&fit=bounds)






































