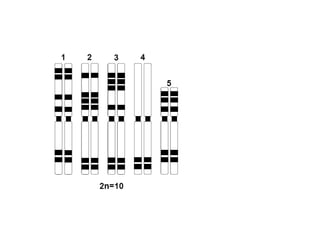
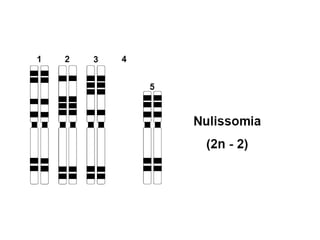
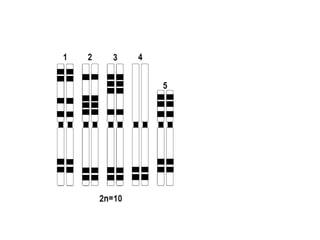
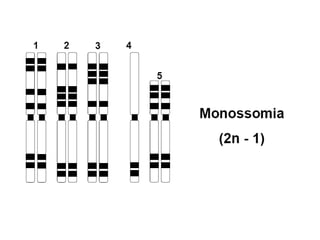
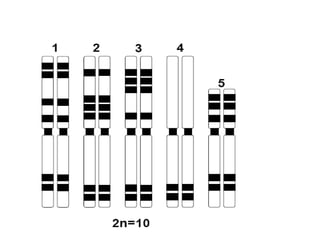
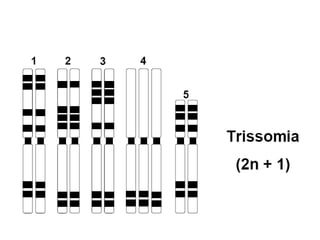
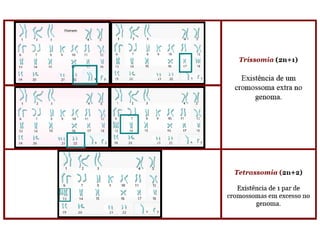
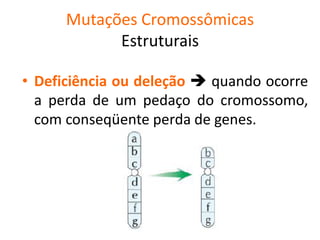
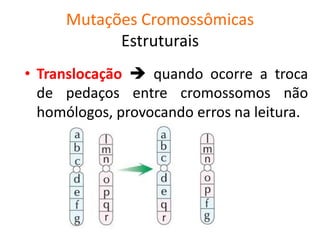
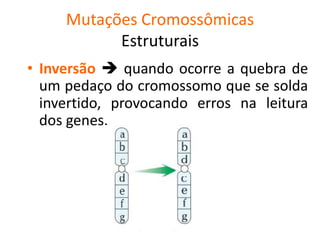
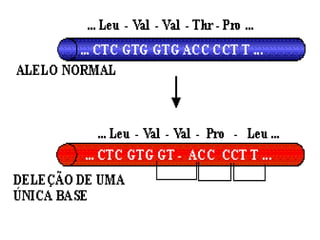
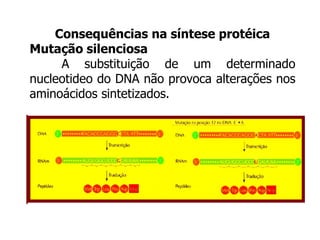
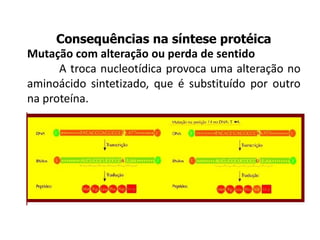
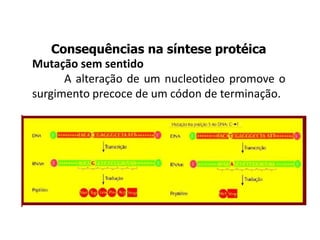
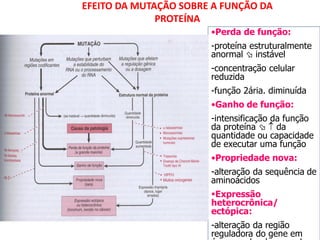
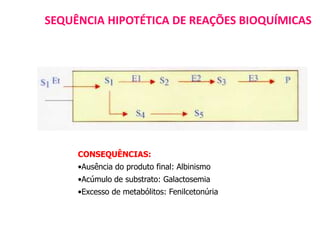
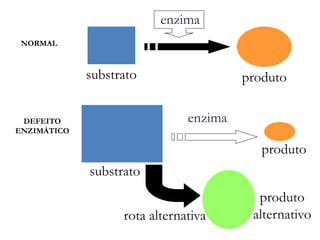
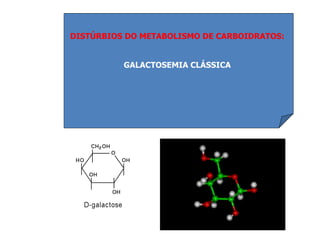
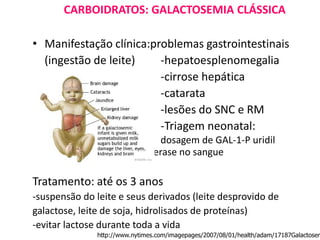
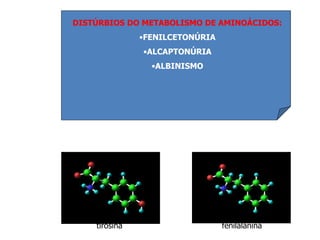
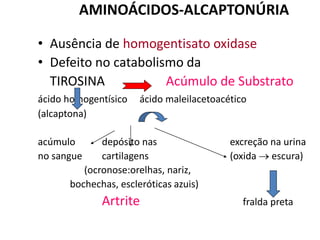
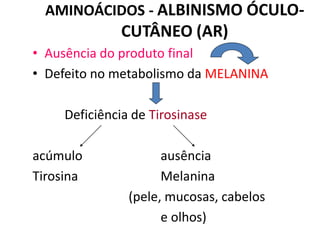
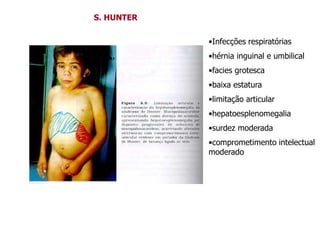
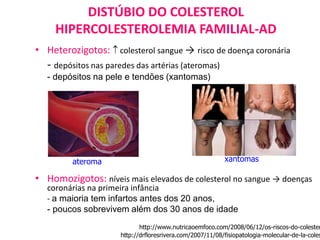
O documento discute mutações no DNA, definindo-as como alterações ou modificações súbitas em genes ou cromossomos que podem acarretar variação hereditária. Descreve tipos de mutações como cromossômicas, gênicas e genômicas e seus efeitos, e discute doenças causadas por mutações como fenilcetonúria, alcaptonúria e doenças de armazenamento lisossômico.