


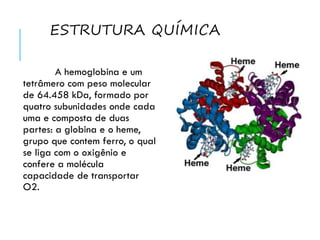
![O GRUPO HEME
O grupo prostético
Heme confere as proteínas a
que está ligado uma cor
característica, e é constituído
por uma parte orgânica e um
átomo de ferro, no estado
ferroso [Fe(II)].](https://image.slidesharecdn.com/hemoglobinopatiasversosemvdeos-140521015634-phpapp01/85/Hemoglobinopatias-4-320.jpg)


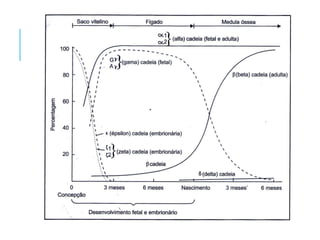





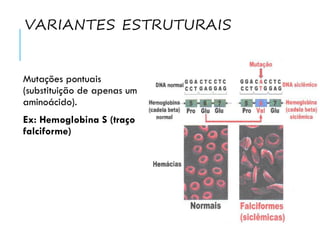






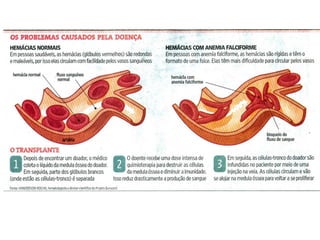

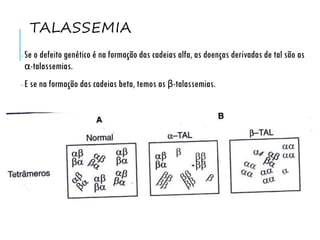
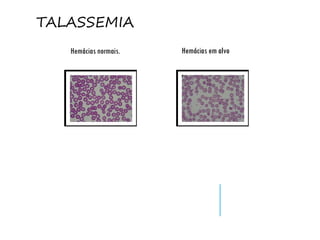












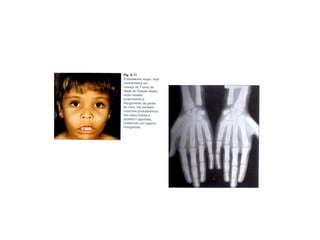



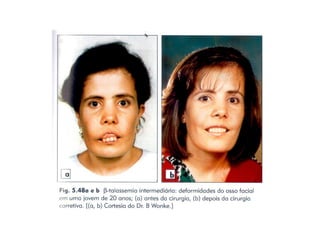
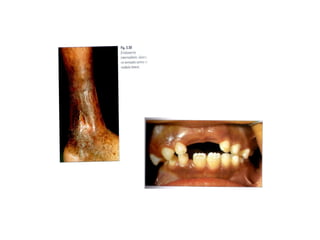

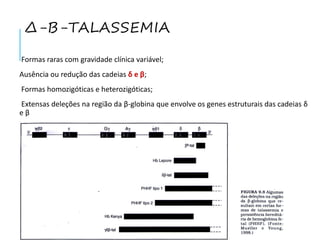






O documento discute doenças relacionadas à hemoglobina, incluindo a estrutura e função da hemoglobina, tipos de hemoglobina em diferentes estágios de desenvolvimento, variantes normais e anormais da hemoglobina, e doenças genéticas como talassemia e anemia falciforme.

































![Anemia%20 reduzida%20[modo%20de%20compatibilidade]](https://cdn.slidesharecdn.com/ss_thumbnails/anemia20reduzida20modo20de20compatibilidade-130519081516-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)






















