




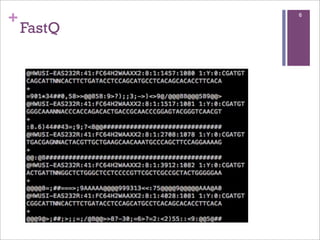







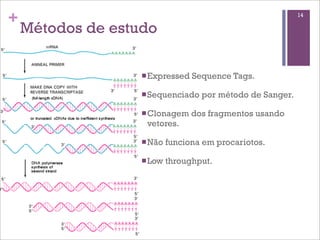
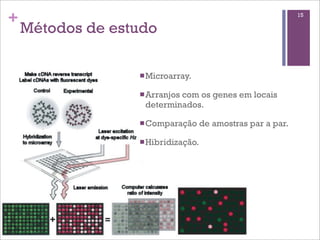












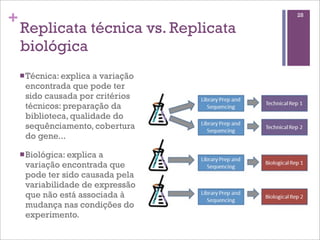

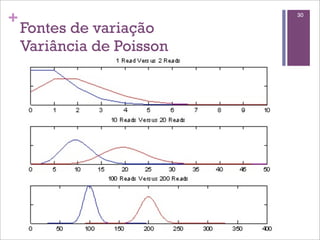

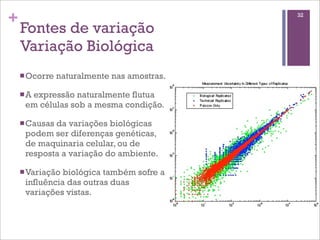






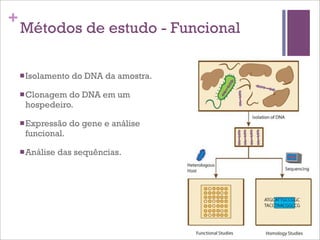


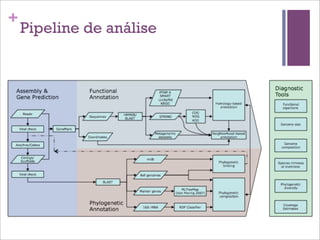

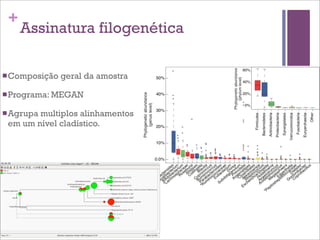




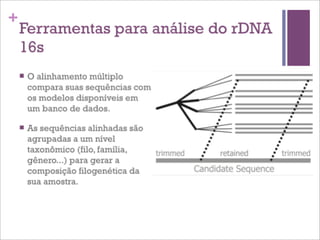





Este documento discute estratégias de sequenciamento, análise de sequências e aplicações de genômica, transcritômica e metagenômica. Aborda tópicos como sequenciadores, formatos de arquivos, montagem, predição de genes, RNA-seq, análise diferencial de expressão genética e estudos funcionais e filogenéticos em metagenômica.


































































