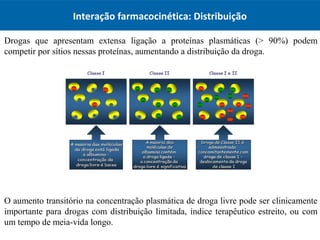
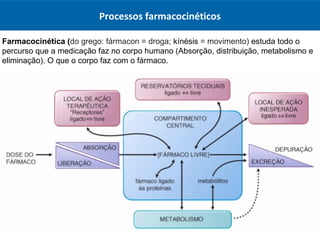
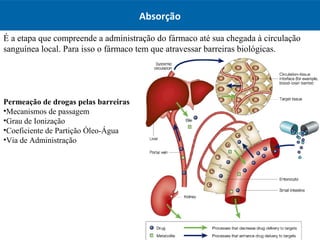
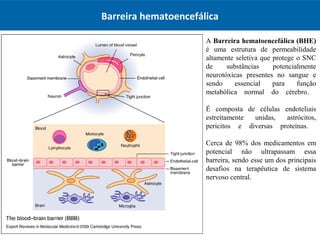
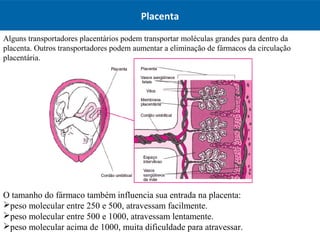
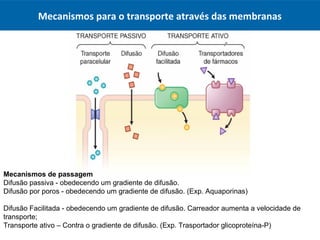
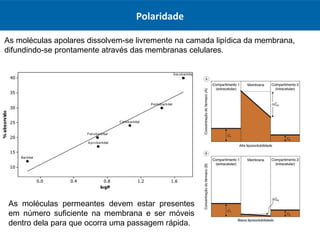
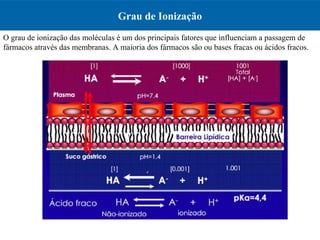
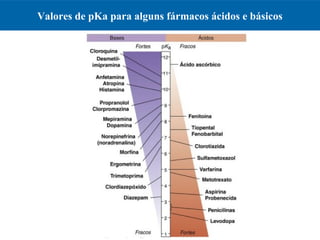
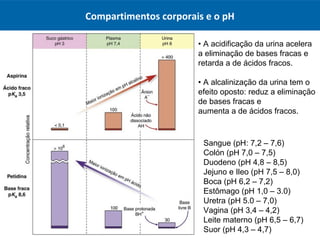
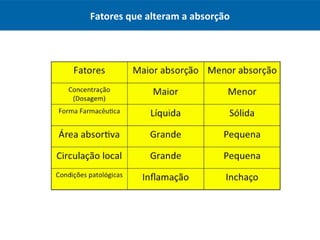
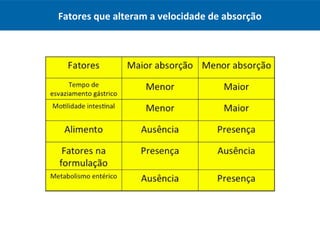
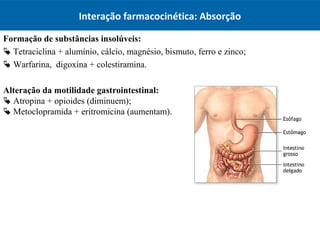
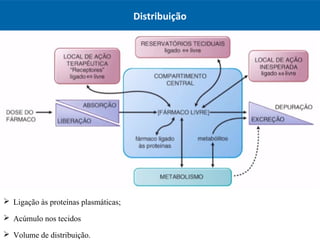
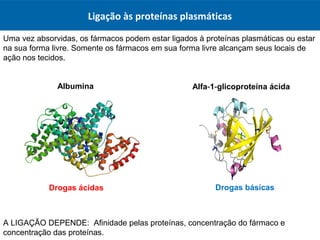
O documento resume os principais conceitos de absorção e distribuição de fármacos. Em particular, discute como fatores como a polaridade, grau de ionização, peso molecular e pH afetam a absorção de fármacos através de membranas biológicas. Também explica como a ligação a proteínas plasmáticas e o volume de distribuição determinam a distribuição de fármacos nos tecidos.








![Biodisponibilidade (F)
Fração inalterada do fármaco que alcança a circulação sistêmica após a administração
por qualquer via.
A concentração do fármaco no sangue em função do tempo é uma medida comum
da extensão da biodisponibilidade de um Fármaco
Biodisponibilidade (F) = [ ] sérica (Cp)
Tempo (h)
v.o.
i.v.](https://image.slidesharecdn.com/aula-basica-adsorcao-distribuicao-180321042853/85/Aula-Basica-Adsorcao-Distribuicao-21-320.jpg)




![Volume de distribuição: Modelo de compartimento único
A equação de Vd, considera o corpo como um compartimento único. E a depuração do Fármaco
nesse modelo segue uma cinética de primeira ordem (exponencial).
Vd = dose
[F]
Modelo aplicado à
maioria das
situações clínicas
Esse modelo é aplicável quando a concentração plasmática cai exponencialmente
após a administração do fármaco.](https://image.slidesharecdn.com/aula-basica-adsorcao-distribuicao-180321042853/85/Aula-Basica-Adsorcao-Distribuicao-31-320.jpg)
![Vd = dose
[F]
Esse volume se refere ao volume de líquido que seria necessário para conter
todo o Fármaco presente no corpo.
Volume de distribuição aparente (Vd)
É um conceito aparente que dá informação sobre como a droga é distribuída no corpo.
SE o Vd é aparente, um valor não real, qual a importância da sua determinação?
O maior determinante é A RELATIVA FORÇA DE LIGAÇÃO DOS FÁRMACOS
AOS COMPONENTES TECIDUAIS COMPARADO COM AS PROTEÍNAS
PLASMÁTICAS](https://image.slidesharecdn.com/aula-basica-adsorcao-distribuicao-180321042853/85/Aula-Basica-Adsorcao-Distribuicao-33-320.jpg)
![Volume de distribuição aparente (Vd)
Fármaco com > Vd:
> [F] nos tecidos
extravasculares
< [F] no sangue
Fármaco com < Vd:
> [F] no sangue
Drogas muito lipossolúveis:
Vd alto
Drogas Hidrossolúveis: Vd
baixo](https://image.slidesharecdn.com/aula-basica-adsorcao-distribuicao-180321042853/85/Aula-Basica-Adsorcao-Distribuicao-34-320.jpg)

![O volume de distribuição aparente pode ser utilizado para calcular a dose de ataque de
alguns fármacos.
Dose de ataque
Dose de ataque = [F] desejada x Vd](https://image.slidesharecdn.com/aula-basica-adsorcao-distribuicao-180321042853/85/Aula-Basica-Adsorcao-Distribuicao-36-320.jpg)