Baixado 712 vezes





![Princípios gerais 24 Março 2009 Farmacocinética [FÁRMACO LIVRE] DOSE DO FÁRMACO Absorção Liberação Metabolização Excreção Metabolização [METABÓLITOS ATIVOS] [METABÓLITOS INATIVOS] “ Corpora non agunt nisi fixata” – Paul Ehrlich LOCAL DE AÇÃO TERAPÊUTICA Reservatórios (tecidos) Local de ação inesperada](https://image.slidesharecdn.com/farmacocintica-090324112037-phpapp02/85/FarmacocineTica-6-320.jpg)








![Eliminação na primeira passagem CL: taxa de eliminação/[droga] Q: fluxo sanguíneo hepático (~90 l/h) 24 Março 2009 Farmacocinética O efeito da eliminação hepática de primeira passagem sobre a biodisponibilidade é expresso como uma RAZÃO DE EXTRAÇÃO:](https://image.slidesharecdn.com/farmacocintica-090324112037-phpapp02/85/FarmacocineTica-21-320.jpg)












![Concentração no equilíbrio Drogas que são administradas em doses múltiplas e contínuas acumulam-se gradativamente no organismo, alcançando um platô de concentração (f = ER) O acúmulo da droga até o ponto em que o equilíbrio é alcançado é um processo cinético de primeira ordem , porque a [ss] é atingida quando a taxa de absorção se iguala à velocidade de eliminação, e os processos envolvidos na cinética não são saturados. O tempo necessário para que a [ss] seja atingida é uma função da t 1/2 da droga t [ss] = 4(t 1/2 ) 24 Março 2009 Farmacocinética](https://image.slidesharecdn.com/farmacocintica-090324112037-phpapp02/85/FarmacocineTica-38-320.jpg)


![Uma aplicação clínica A meia-vida de eliminação de uma droga X é de 10 horas e ela é administrada a cada 8 horas. A [ss] será atingida depois de 40 ou 50 horas. Para que a [ss] seja atingida após 40-50 hs, os intervalos entre as doses devem ser menores do que 1.4x10 1 horas. 8 < 14; Q.E.D A efetividade de um novo esquema posológico só pode ser completamente avaliada depois que outras 4 ou 5 meias-vidas tenham se passado, porque uma nova [ss] deve ser atingida. 24 Março 2009 Farmacocinética](https://image.slidesharecdn.com/farmacocintica-090324112037-phpapp02/85/FarmacocineTica-41-320.jpg)


![http://www.slideshare.net/caio_maximino/farmacocinetica [email_address] 24 Março 2009 Farmacocinética](https://image.slidesharecdn.com/farmacocintica-090324112037-phpapp02/85/FarmacocineTica-46-320.jpg)

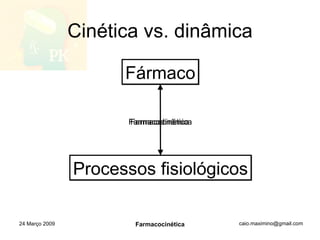
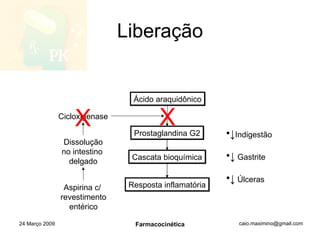
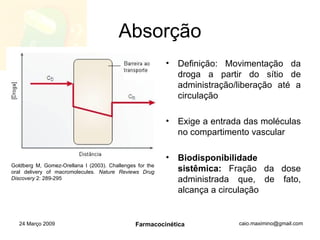
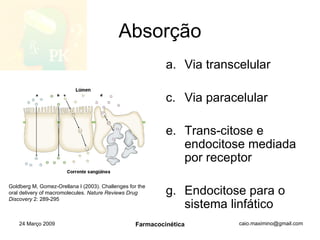
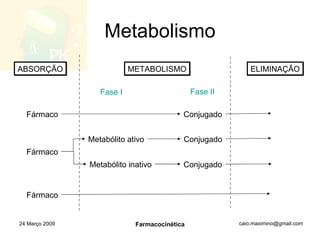
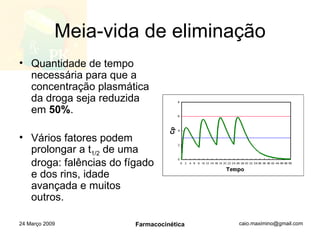
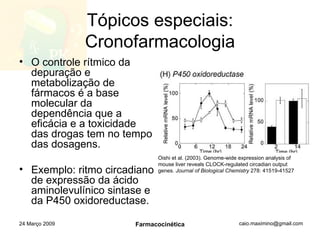
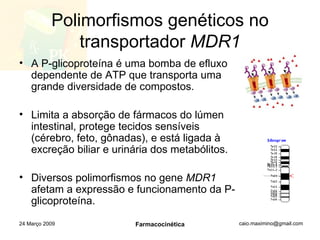
O documento discute os principais conceitos de farmacocinética, incluindo a liberação, absorção, distribuição, metabolismo e excreção de fármacos no corpo. Também aborda como fatores como pH, ligação a proteínas e metabolismo hepático de primeira passagem afetam a biodisponibilidade sistêmica de fármacos administrados por via oral.










































































































