

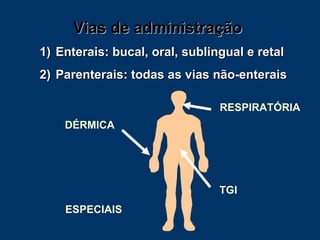

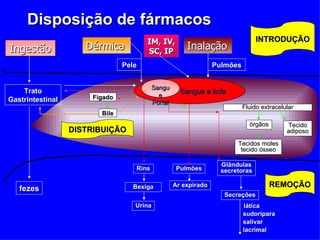
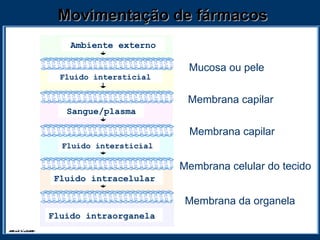
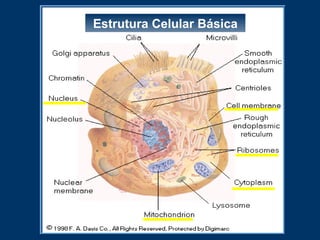
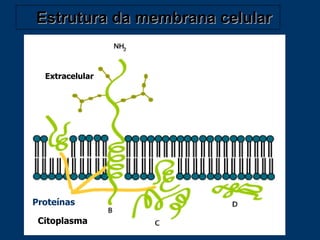



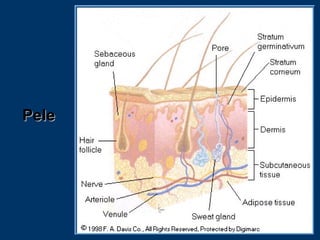

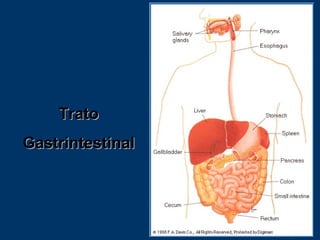

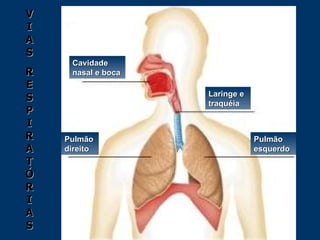







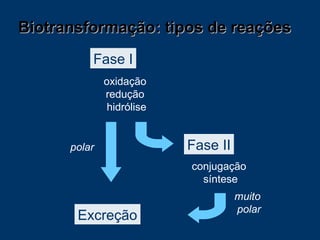


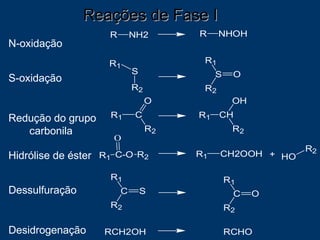
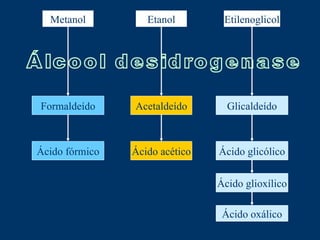



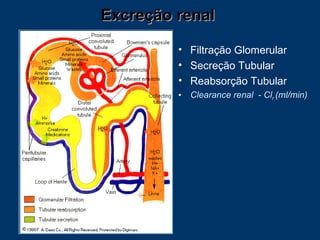
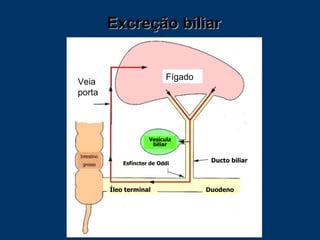
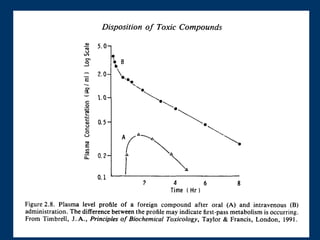
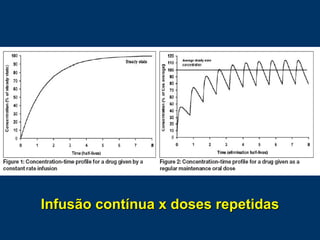
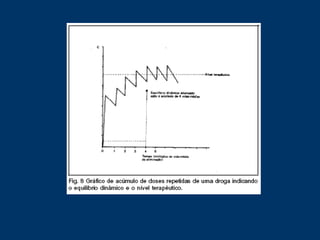
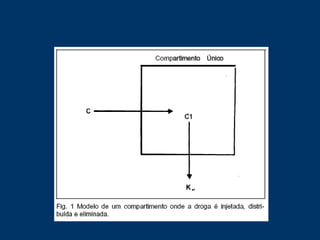



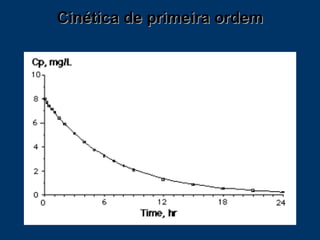
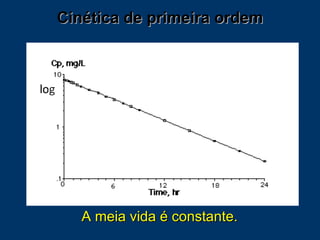



O documento descreve os processos farmacocinéticos de absorção, distribuição, biotransformação e excreção de fármacos no organismo. Detalha os fatores que influenciam cada processo e os mecanismos de transporte através das membranas celulares e barreiras do corpo. Também explica os sistemas enzimáticos envolvidos na biotransformação hepática de primeira e segunda fase e os parâmetros farmacocinéticos utilizados para quantificar esses processos.









































































