Baixado 64 vezes




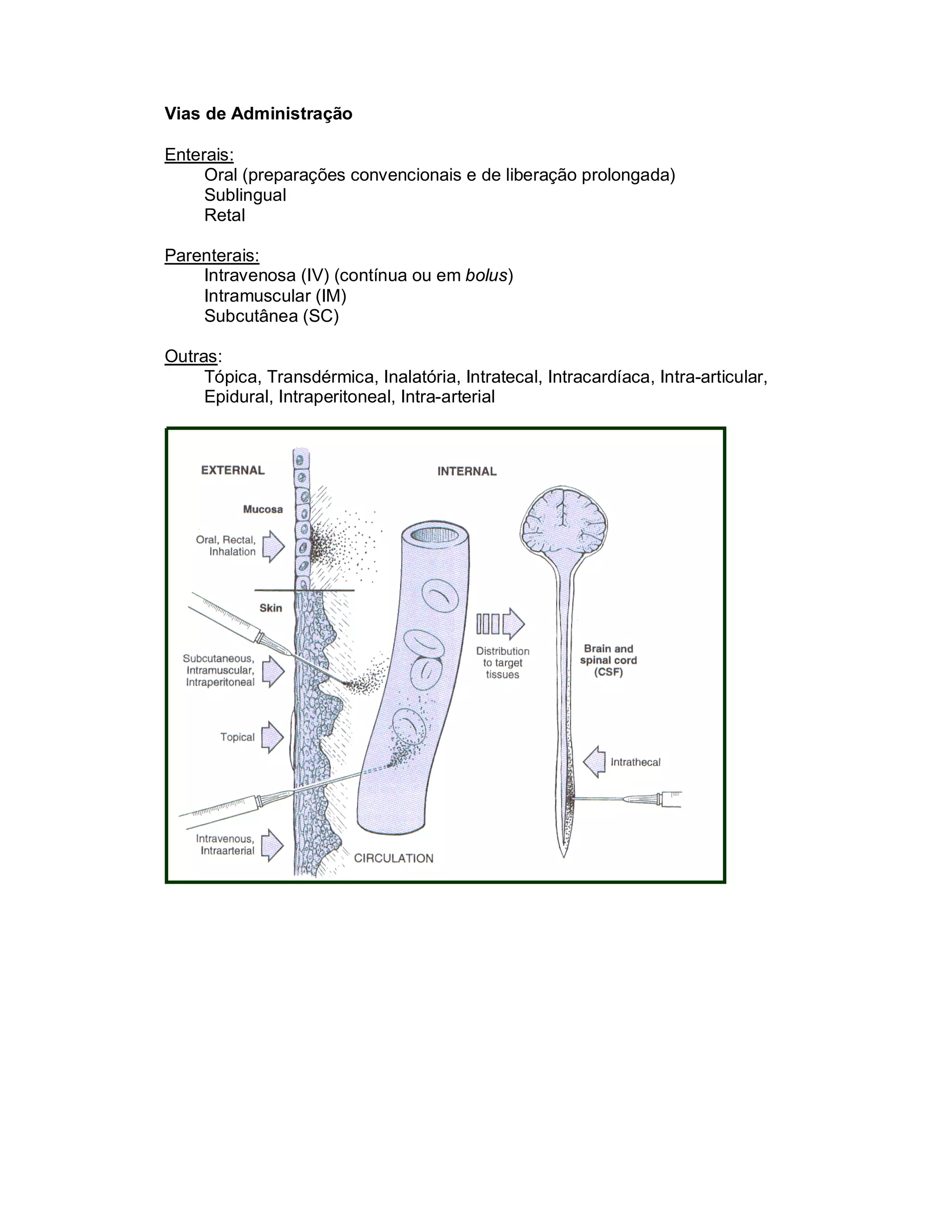
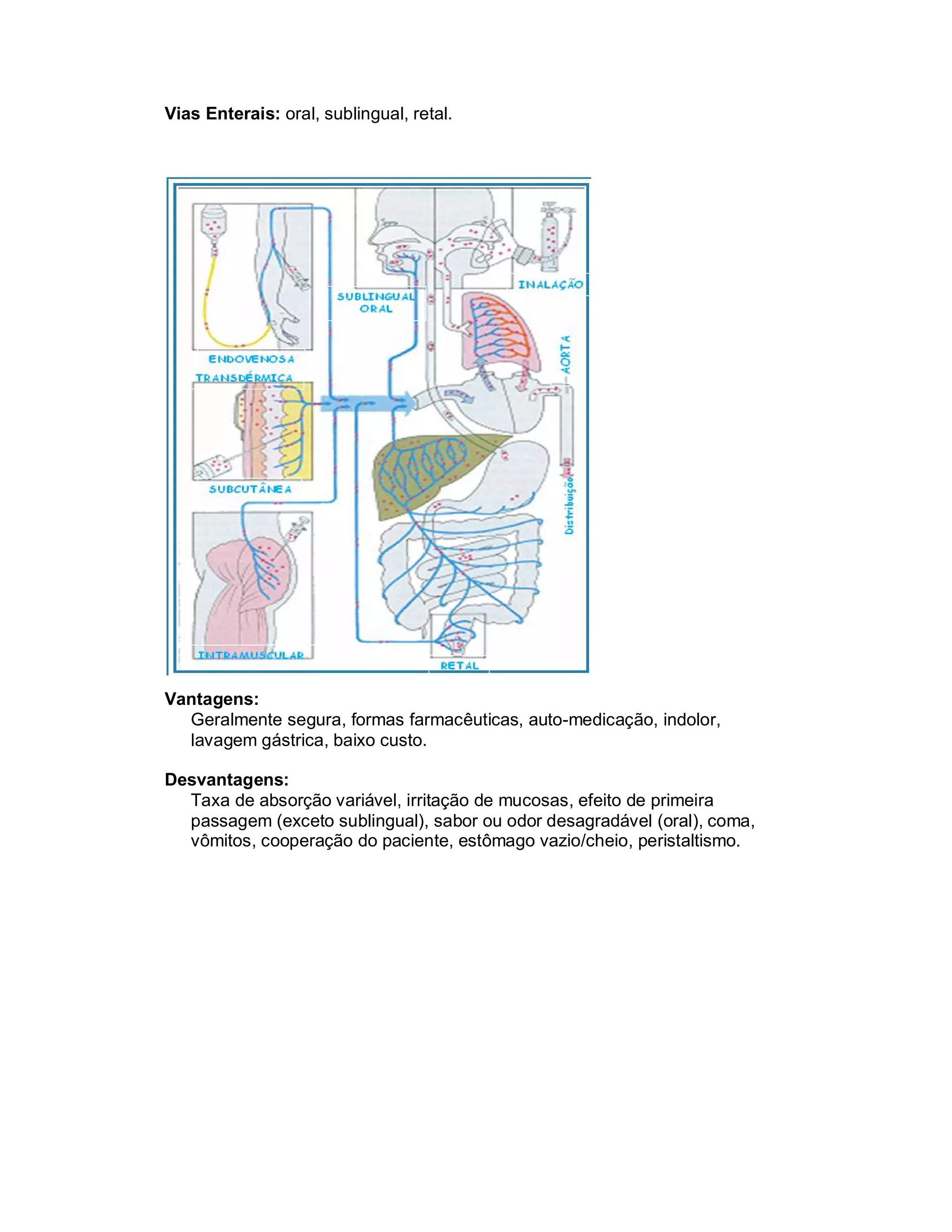

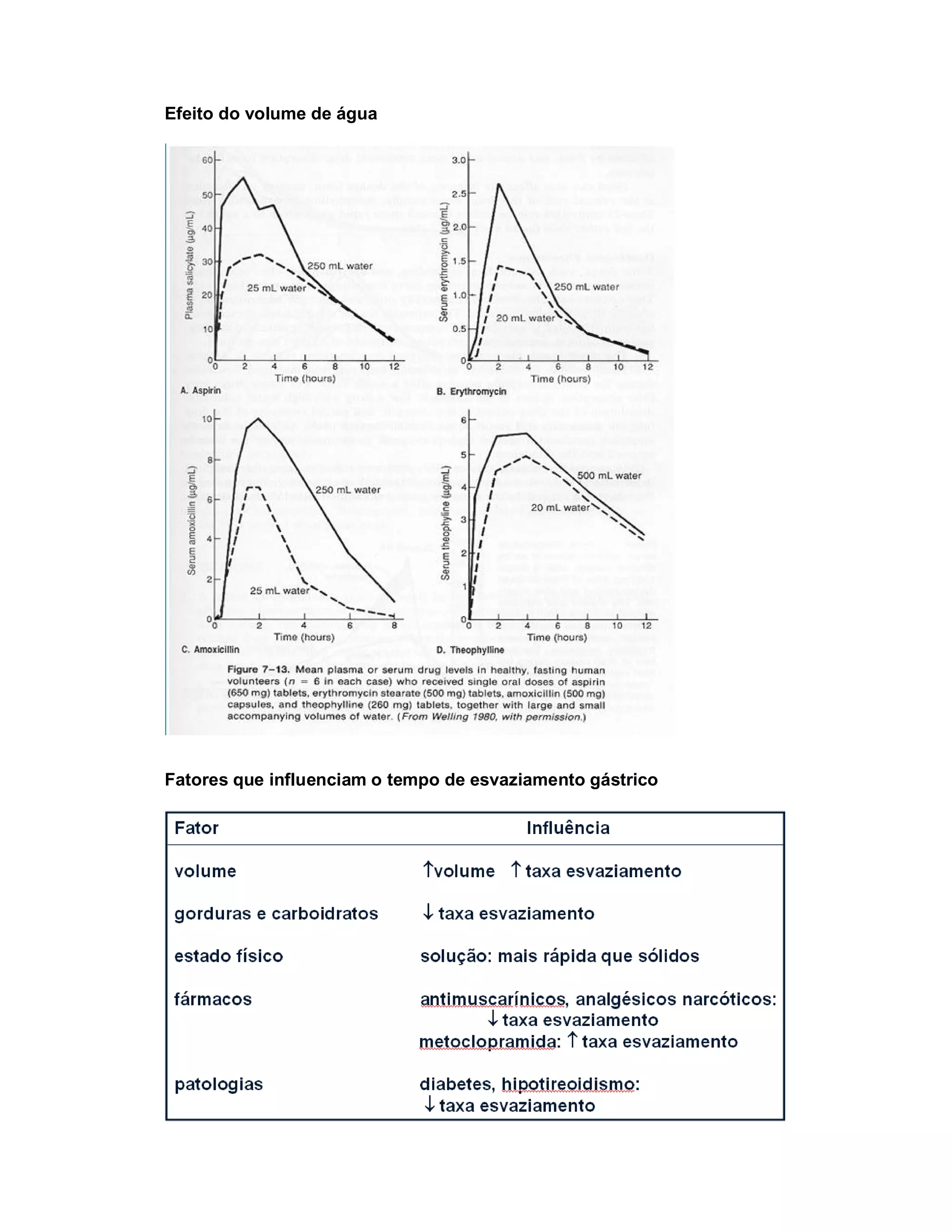









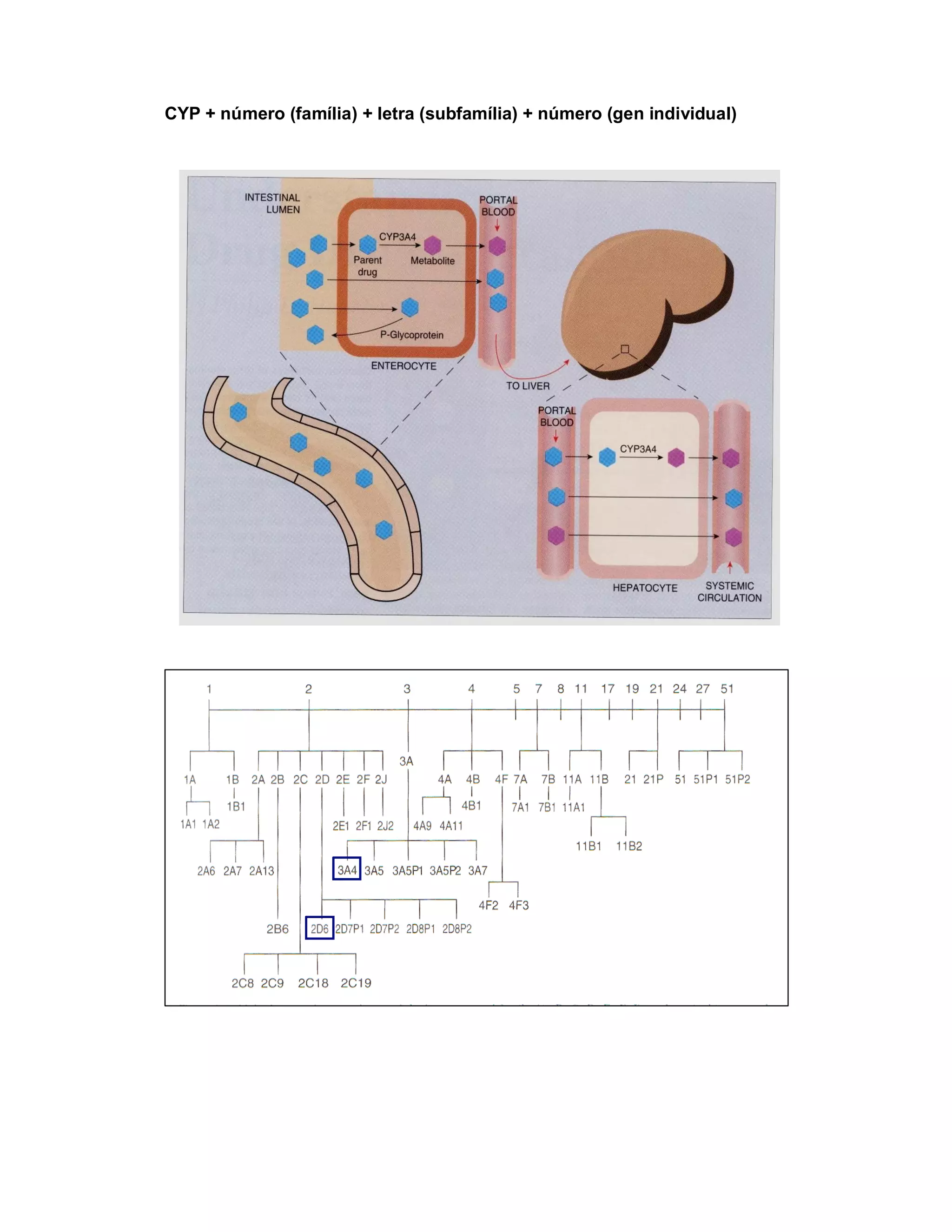



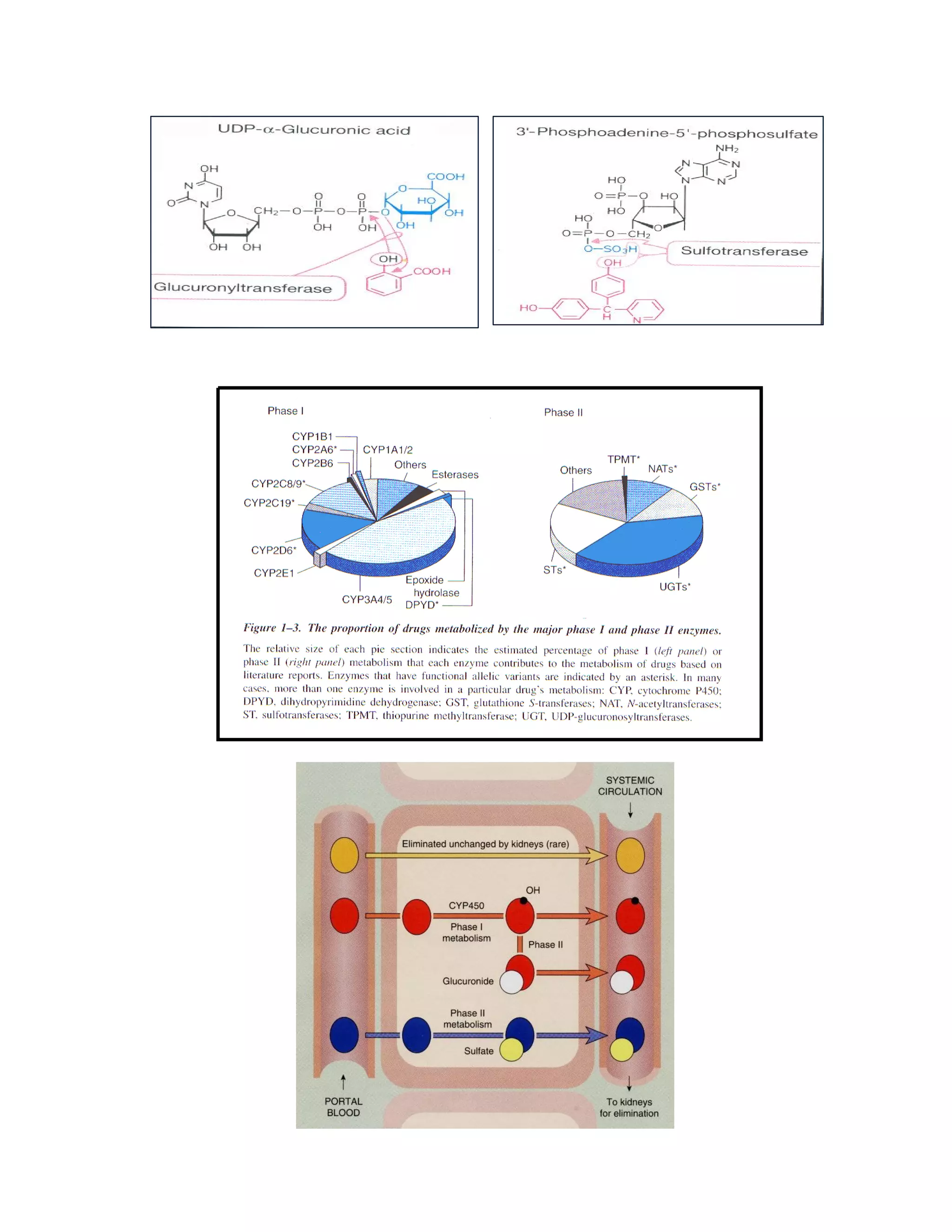




Este documento discute os principais conceitos de farmacocinética, incluindo absorção, distribuição, metabolismo e eliminação de fármacos no organismo. Ele explica os diferentes mecanismos de transporte de fármacos através das membranas celulares e barreiras biológicas, assim como os fatores que influenciam a absorção de fármacos via oral e outras vias de administração. O documento também aborda os processos de biotransformação hepática e conjugação de fármacos, assim como os fatores que afet















































