Transferir como PDF, PPTX






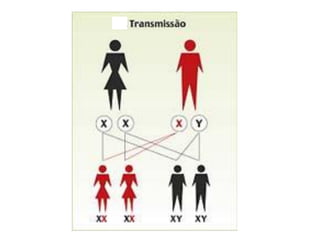

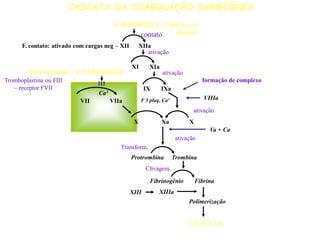


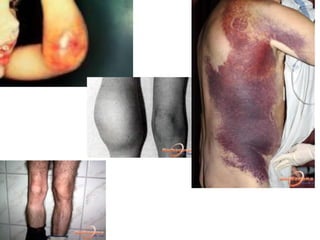

















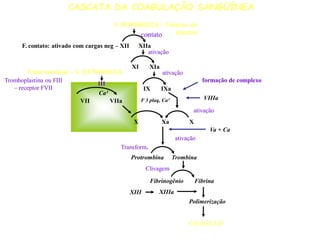






O documento discute distúrbios da coagulação como hemofilias, coagulação intravascular disseminada e trombofilias. Ele descreve as causas, manifestações clínicas e diagnóstico laboratorial destas condições, bem como o tratamento com fatores de coagulação, heparina e anticoagulantes orais.



















































































