Baixado 23 vezes


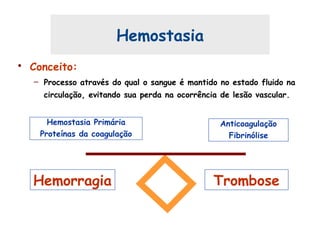
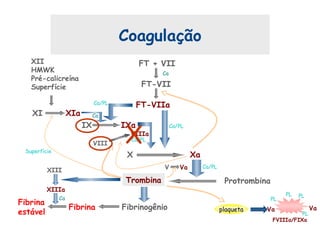

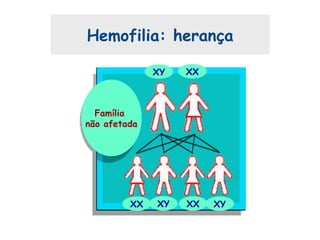
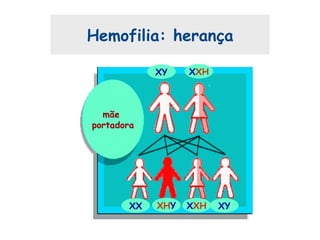
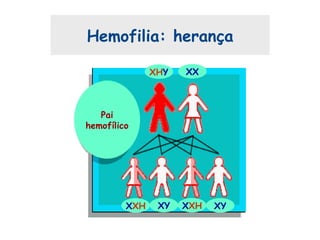
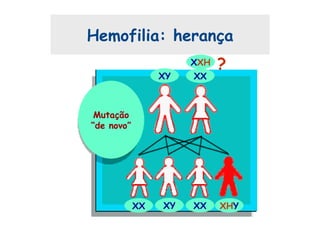
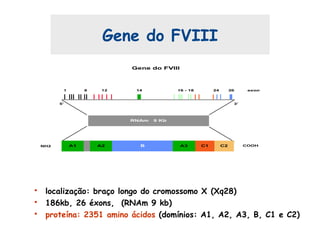
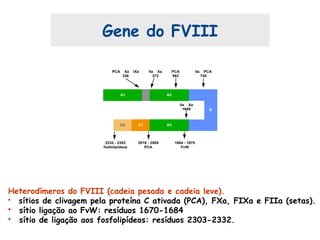

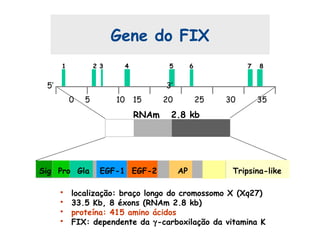



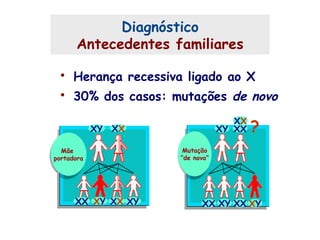

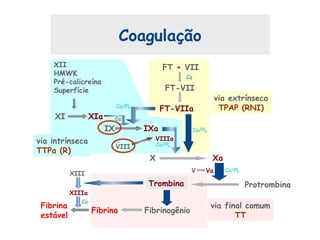



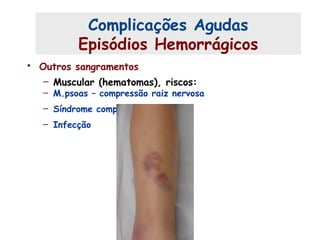



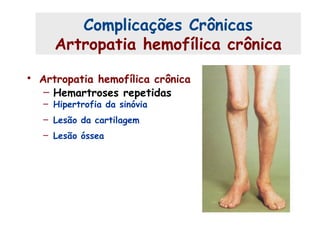
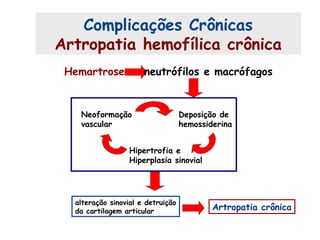
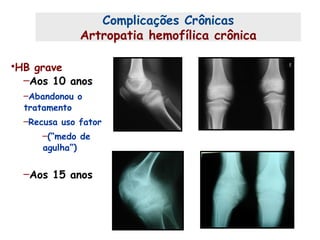
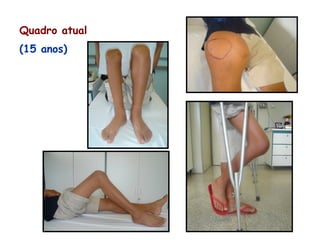




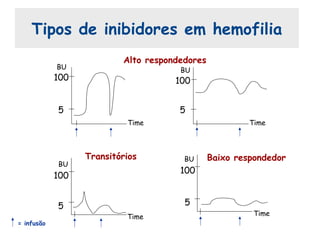




Este documento resume aspectos gerais da hemofilia, incluindo: 1) A hemofilia é causada por deficiência dos fatores VIII ou IX, levando a distúrbios na coagulação do sangue. 2) Ela é hereditária ligada ao cromossomo X e pode ser grave, moderada ou leve dependendo dos níveis de fator. 3) Complicações incluem artropatia, infecções e inibidores contra os fatores da coagulação.





















































