Transferir como PDF, PPTX


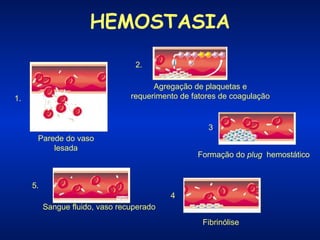
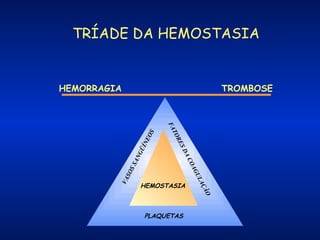
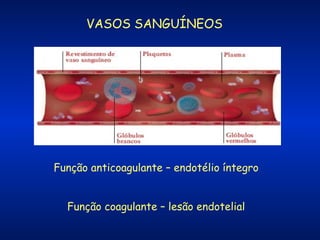


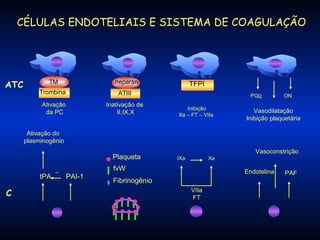

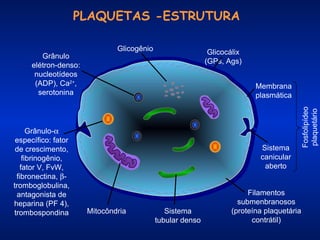

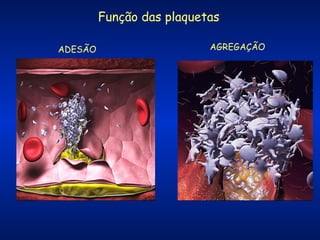

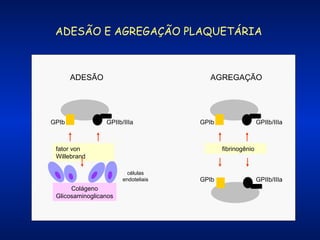
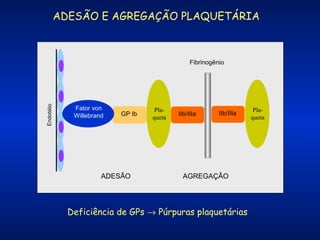
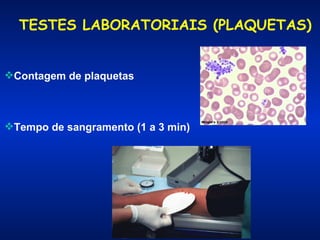

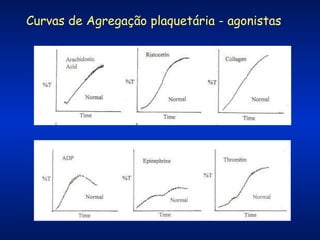


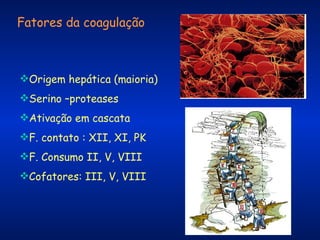
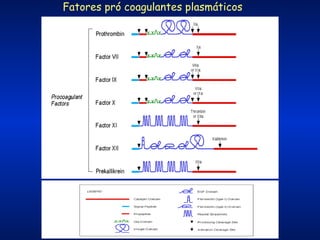
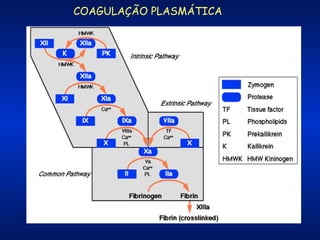
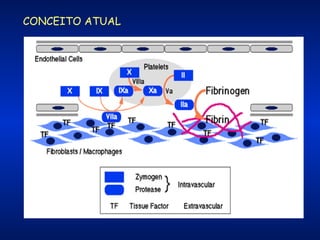
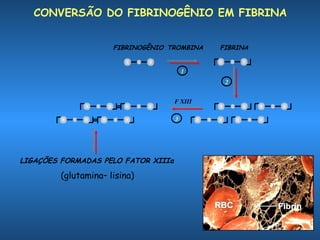
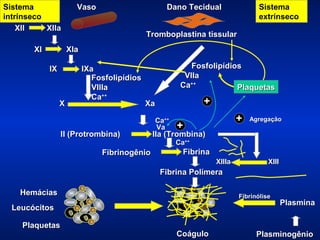
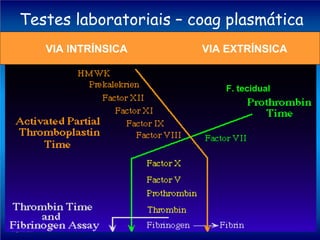
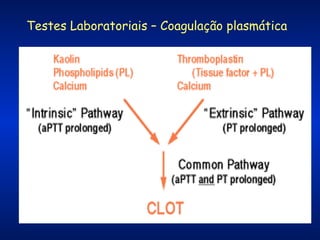












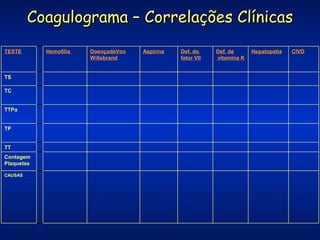
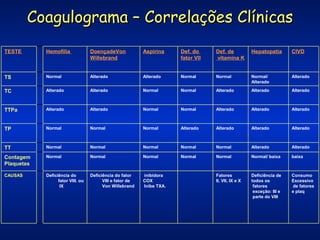

O documento descreve os processos da hemostasia, incluindo as funções dos vasos sanguíneos, plaquetas e fatores de coagulação. Discutem-se os mecanismos de adesão e agregação plaquetária, ativação da cascata de coagulação e formação do coágulo. Também são apresentados testes laboratoriais relevantes para avaliar a hemostasia.



































































