Baixado 82 vezes
![Coagulação sanguínea
Origem: Wikipédia, a enciclopédia livre.
A coagulação sanguínea é uma sequência complexa de reações químicas que resultam na
formação de um coágulo de fibrina. É uma parte importante da hemostasia (o cessamento da perda
de sangue de um vaso danificado), na qual a parede de vaso sanguíneo danificado é coberta por um
coágulo de fibrina para parar o sangramento e ajudar a reparar o tecido danificado. Desordens na
coagulação podem levar a um aumento no risco de hemorragia, trombose ou embolismo.
A coagulação é semelhante nas várias espécies de mamíferos. Em todos eles o processo envolve
um mecanismo combinado de células (plaquetas) e proteínas (fatores de coagulação). Esse sistema
nos humanos é o mais extensamente pesquisado e consequentemente o mais bem conhecido. Esse
artigo é focado na coagulação sanguínea humana.
Índice
[esconder]
1 Visão geral
2 Hemostasia Primária
3 Hemostasia Secundária
o 3.1 Cascata de Coagulação
3.1.1 Vía intrínseca
3.1.2 Vía Extrínseca
3.1.3 Formação da Trombina
o 3.2 Co-fatores da coagulação
o 3.3 Inibidores da Coagulação
4 Distúrbios da hemostasia
5 Fatores de Coagulação
6 Fibrinólise
7 História
o 7.1 Descobertas iniciais
o 7.2 Fatores de coagulação
o 7.3 Nomenclatura
8 Bibliografia
9 Referências
10 Ligações externas
[editar]Visão geral](https://image.slidesharecdn.com/coagulaosangunea-120527102739-phpapp01/85/Coagulacao-sanguinea-1-320.jpg)
![Em um indivíduo normal, a coagulação é iniciada dentro de 20 segundos após a lesão ocorrer ao
vaso sanguíneo causando dano às células endoteliais. As plaquetas formam imediatamente um
tampão plaquetário no local da lesão. Essa é a chamada hemostasia primária. A hemostasia
secundária acontece quando os componentes do plasmachamados fatores de
coagulação respondem (em uma completa cascata de reações) para formar fios de fibrina, que
fortalecem o tampão plaquetário. Ao contrário da crença comum, a coagulação a partir de um corte
na pele não é iniciada pelo ar ou através da secagem da área, na verdade ocorre através das
plaquetas que se aderem e que são ativadas pelo colágeno do endotélio do vaso sanguíneo que fica
exposto, quando cortado o vaso. As plaquetas ativadas então liberam o conteúdo de seus grânulos,
que contém uma grande variedade de substâncias que estimulam uma ativação ainda maior de
outras plaquetas e melhoram o processo hemostásico..
[editar]Hemostasia Primária
Vasoconstrição: primeiramente o vaso lesado se contrai.
Adesão: Inicia-se quando as plaquetas se aderem ao endotélio vascular. Essa aderência
acontece com uma ligação entre a glicoproteína Ib/IX/V na superfície das plaquetas e colágeno
exposto durante a lesão do endotélio. Essa ligação é mediada pelo fator de von Willebrand que
funciona como uma "ponte" entre a superfície da plaqueta e o colágeno. Quando ocorre uma
desordem qualitativa ou quantitativa deste fator ocorre a Doença de von Willebrand. A
aderência leva a ativação plaquetária. Quando ocorre falta da glicoproteína Ib ocorre
a Síndrome de Bernard-Soulier.
Ativação Plaquetária: Quando ocorre a ativação das plaquetas, elas mudam de forma e liberam
conteúdos dos seus grânulos no plasma entre eles produtos de oxidação do ácido araquidônico
pela via cicloxigenase (PGH2 e seu produto, o tromboxane), ADP, fator de ativação plaquetária
(PAF). Quando ocorre uso de aspirina por um indivíduo, ocorre a inativação da enzima
cicloxigenase evitando a síntese de PGH2 e tromboxane e ocorre um prolongamento do tempo
de sangramento.
Agregação plaquetária: as plaquetas se agregam uma às outras, formando o chamado "trombo
branco".
[editar]Hemostasia Secundária
[editar]Cascata de Coagulação](https://image.slidesharecdn.com/coagulaosangunea-120527102739-phpapp01/85/Coagulacao-sanguinea-2-320.jpg)
![Cascata de Coagulação. Legenda: HMWK =Cininogênio de Alto Peso Molecular, PK= Precalicreína, TFPI
=Inibidor do Fator Tissular. Seta preta= conversão/ativação de fatores. Seta vermelha=ação dos inibidores. Seta
Azul= reações catalisadas por fatores ativados. Seta Cinza= várias funções da trombina.
Possui duas vías: intrínseca (vía da ativação de contato) e extrínseca (vía do fator tissular). Ambas
vías tem grande importância e acabam se juntando para formação do coágulo de fibrina. Os fatores
de coagulação são numerados por algarismos romanos e a adição da letra aindica que eles estão
em sua forma ativada. Os fatores de coagulação são geralmente enzimas (serino proteases) com
exceção dos fatores V e VIII que são glicoproteínas e do fator XIII que é uma transglutaminase. As
serino proteases agem clivando outras proteínas.
[editar]Vía intrínseca
Necessita dos factores de coagulação VIII, IX, X, XI e XII além das proteínas pré-calicreína
(PK), cininogênio de alto peso molecular (HWHK) e íons cálcio.
Começa quando a PK, o HWHK, factor XI e XII são expostos a cargas negativas do vaso
lesado, isso é chamado de "fase de contacto".
A pré-calicreína então converte-se em calicreína e esta activa o fator XII.
O fator XII activado acaba convertendo mais prekalicreína em calicreína e activando o factor XI.
Na presença de íos cálcio, o fator XI ativado ativa o IX. Por sua vez o factor IX activado junto
com o factor VIII activado, levam à activação do factor X. Deste modo, o complexo enzimático](https://image.slidesharecdn.com/coagulaosangunea-120527102739-phpapp01/85/Coagulacao-sanguinea-3-320.jpg)
![constituído pelo factor X activado, juntamente com o factor V activado e Ca++, denomina-se de
Protrombinase.
[editar]Vía Extrínseca
Após a lesão vascular, o fator tecidual (fator III) é lançado e forma junto ao fator VII ativado um
complexo (Complexo FT-FVIIa) que irá ativar os fatores IX e X.
O fator X ativado junto ao fator V ativado formam um complexo (Complexo protrombinase) que
irá ativar a protrombina em trombina.
A trombina ativa outros componentes da coagulação entre eles os fatores V e VII (que ativa o
fator XI que por sua vez ativa o fator IX). Os fatores VII, juntamente com o factor tecidual e
Ca++ ativados formam o Complexo Tenase Extrínseco que por sua vez ativa o fator X.
[editar]Formação da Trombina
O ponto comum entre as duas vías é a ativação do fator X em fator Xa. Por sua vez, o Fator Xa
converte a protrombina em trombina. A trombina tem várias funções:
A principal é a conversão do fibrinogênio em fibrina. O fibrinogênio é uma molécula constituída
por dois pares de três cadeias diferentes de polipeptídeos. A trombina converte o fibrinogênio
em monômeros de fibrina e ativa o fator XIII. Por sua vez, o fator XIIIa liga de forma cruzada a
fibrina à fibronectina e esta ao colágeno.
Ativação dos fatores VIII e V e seus inibidores, a proteína C (na presença de trombomodulina).
Ativação do Fator XIII.
[editar]Co-fatores da coagulação
Cálcio: Age mediando a ligação do Fatores IXa e Xa junto as plaquetas através da ligação
terminal dos resíduos gamma-carboxi dos fatores IXa e Xa junto a fosfolípideos da membrana
das plaquetas. O cálcio também está presente em vários pontos da cascata da coagulação.
Vitamina K: Adiciona um carboxil ao ácido glutâmico residual dos fatores II, VII, IX e X e
também as proteínas C, S e Z.
[editar]Inibidores da Coagulação
Três substâncias mantêm a cascata da coagulação em equilíbrio. Defeitos quantitativos e
qualitativos destas susbstâncias podem aumentar a tendência a trombose.
Proteína C: Age degradando os fators Va e VIIIa. É ativado pela trombina em presença da
trombomodulina e da co-enzima proteína S.
Antitrombina: Age degradando as serino proteases (trombina, FXa, FXIIa e FIXa)
Inibidor do Fator Tissular: Inibe o FVIIa relacionado com a ativação do FIX e FX.
Exemplos de anticoagulantes farmacológicos:](https://image.slidesharecdn.com/coagulaosangunea-120527102739-phpapp01/85/Coagulacao-sanguinea-4-320.jpg)
![ Heparina
Varfarina
[editar]Distúrbios da hemostasia
Distúrbios das plaquetas e da parede do vaso
Púrpura trombocitopênica imune (ITP)
Púrpura trombocitopênica trombótica (TTP)
Síndrome hemolítica-urêmica (HUS)
Trombastenia de Glanzmann
Síndrome de Bernard-Soulier (complexo gricoprotéico Ib-IX-V anormal)
Storage pool disorders
Hemoglobinúria paroxística noturna
Síndrome da plaqueta cinza: deficiência de grânulos alfa.
Síndrome de Hermansky-Pudlak: deficiência de grânulos densos.
Distúrbios da coagulação e trombose
Coagulação intravascular disseminada
Deficiências de fatores
Hemofilia A (deficiência do Fator VIII)
Hemofilia B (deficiência de Fator IX, "Christmas disease")
Hemofilia C (deficiência de Fator XI, tendência de sangramento suave)
Doença de Von Willebrand (o distúrbio de sangramento mais comum)
Inibidores de fator
Disfunção plaquetária
Distúrbios de predisposição a trombose (veja: hipercoagulabilidade)
Trombocitopenia induzida por heparina e trombose ("síndrome do coágulo branco")
Síndrome antifosfolípide
Lúpus anticoagulante
Anticorpo anticardiolipina
Fator V de Leiden e resistência à proteína C ativada
Mutação da protrombina
Deficiência de proteína C
Deficiência de proteína S
Deficiência de antitrombina III
Níveis aumentados anormalmente dos fatores VIII e XI
[editar]Fatores de Coagulação](https://image.slidesharecdn.com/coagulaosangunea-120527102739-phpapp01/85/Coagulacao-sanguinea-5-320.jpg)
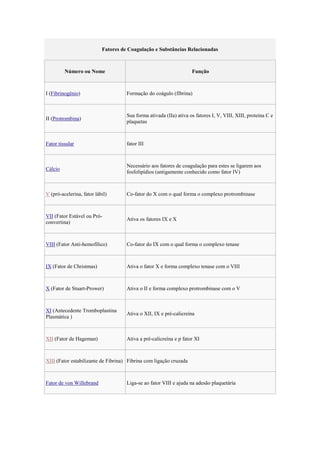
![Ativa o XII e a pré-calicreína. Cliva o clininogênio de alto peso
Pré-calicreína
molecular.
Cininogênio de alto peso
Ajuda na ativação do XII, XI, e pré-calicreína
molecular (HMWK)
Fibronectina Ajuda na adesão celular
Antitrombina III Inibe o fator IIa, Xa, e outras proteases;
Co-fator heparina II Inibe o IIa, co-fator para heparina
Proteína C Inativa o Va e VIIIa
Proteína S Co-fator para ativação da proteína C
Ajuda na adesão da trombina aos fosfolipideos e estimula a
Proteína Z
degradação do fator X pelo ZPI
Proteína Z-relacionada ao inibidor Degrada fatores X (na presença da proteína Z) e XI
de protease (ZPI) (independentemente)
Plasminogênio Converte-se em plasmina, lisa a fibrina e outras proteínas
Alfa 2-antiplasmina Inibe a plasmina
Ativador do plasminogênio
Ativa o plasminogênio
tissular (tPA)
Uroquinase Ativa o plasminogênio
[editar]Fibrinólise
É uma resposta ao depósito de fibrina formado no organismo de um indivíduo. O plasminogênio
liberado pelas células endoteliais é ativado em plasmina cuja função é degradar a fibrina formada.
[editar]História](https://image.slidesharecdn.com/coagulaosangunea-120527102739-phpapp01/85/Coagulacao-sanguinea-7-320.jpg)
![[editar]Descobertas iniciais
Teorias sobre a coagulação do sangue já existiam desde a antiguidade, mas foi no século XIX que
as primeiras substâncias químicas foram descobertas. O fisiologista alemãoJohannes Müller (1801-
1858) descreveu a fibrina, a substância que compõem o trombo. Seu precursor solúvel,
o fibrinogênio, foi assim batizado por Rudolf Virchow (1821-1902), e isolado quimicamente
por Prosper Sylvain Denis (1799-1863). Alexander Schmidt sugeriu que a conversão de fibrinogênio
em fibrina é o resultado de uma processo enzimático, e rotulou a enzima hipotética de "trombina" e
seu precursor "protrombina".[1][2] Nicolas Maurice Arthus descobriu em 1890 que o cálcio é essencial
na coagulação.[3][4] As plaquetasforam identificadas em 1865, e sua função foi elucidada por Giulio
Bizzozero em 1882.[5]
A teoria do que a trombina é gerada pela presença de fator tecidual foi consolidada por Paul
Morawitz em 1905.[6] Nesta época, sabia-se que o fator III é liberado pelos tecidos lesados, reagindo
com a protrombina (II), que, juntamente com cálcio (IV), forma a trombina, que por sua vez converte
o fibrinogênio em fibrina (I).[7]
[editar]Fatores de coagulação
O restante dos fatores bioquímicos no processo de coagulação foram amplamente descobertos
no século XX.
Um primeiro indício quanto à própria complexidade do sistema de coagulação foi a descoberta
de proacelerina (mais tarde chamada de Fator V) por Paul Owren (1905-1990) em 1947. Ele
também postula que esta substância era precursora da acelerina (Fator VI), que mais tarde tornou-
se a forma ativada de fator V (ou Va), daí, o VI não está em uso ativo.[7]
O fator VII (também conhecido como acelerador da conversão sérica de
protrombina ou proconvertina, precipitada por sulfato de bário) foi descoberto em um paciente jovem
do sexo feminino em 1949 e 1951 por diferentes grupos.
Factor VIII acabou por ser deficiente no clinicamente reconhecido mas etiologicamente
indescritível hemofilia A, foi identificado em 1950 e é também conhecida como globulinaanti-
hemofílica, devido à sua capacidade de corrigir hemofilia A.[7]
O Fator IX foi descoberto em 1952, em um paciente jovem com hemofilia B chamado Stephen
Christmas (1947-1993). Sua deficiência foi descrito pelo Dr. Rosemary Biggs e pelo professor R.G.
MacFarlane em Oxford, no Reino Unido. O fator é, portanto, chamado Fator de Christmas.
Christmas vive no Canadá, e fez campanha para a segurança das transfusões de sangue, até
sucumbir à transfusão relacionadas com AIDS aos 46 anos. Um nome alternativo para o fator de
plasma é componente tromboplastina, dado por um grupo independente, na Califórnia.[7]
O Fator Hageman, agora conhecido como fator XII, foi identificado em 1955 em um paciente
assintomático com um tempo de sangramento prolongado chamado John Hageman. O fator X, ou
fator Stuart-Prower, foi descoberto no ano seguinte, em 1956. Esta proteína foi identificada na Sra.](https://image.slidesharecdn.com/coagulaosangunea-120527102739-phpapp01/85/Coagulacao-sanguinea-8-320.jpg)
![Audrey Prower de Londres, que tinha tendência ao sangramento ao longo de toda a sua vida. Em
1957, um grupo americano identificou o mesmo fator em Sr. Rufus Stuart. Já os fatores XI e XIII
foram identificados em 1953 e 1961, respectivamente.[7]
A visão de que o processo de coagulação é uma "cascata" ou "cachoeira" foi enunciada quase
simultaneamente por MacFarlane[8] no Reino Unido e por Davie e Ratnoff[9] nosEstados Unidos.](https://image.slidesharecdn.com/coagulaosangunea-120527102739-phpapp01/85/Coagulacao-sanguinea-9-320.jpg)

A coagulação sanguínea é uma sequência complexa de reações químicas que resultam na formação de um coágulo de fibrina. O processo envolve plaquetas, fatores de coagulação e proteínas que atuam em cascata para parar hemorragias. Distúrbios na coagulação podem causar problemas de sangramento ou trombose.



































































