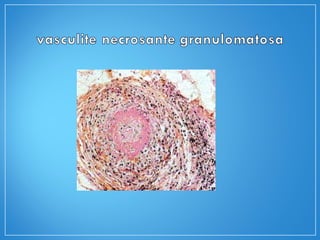
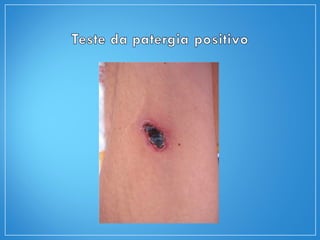
1) O documento descreve as características clínicas, exames complementares e tratamento da granulomatose de Wegener e da arterite de células gigantes, duas vasculites sistêmicas.
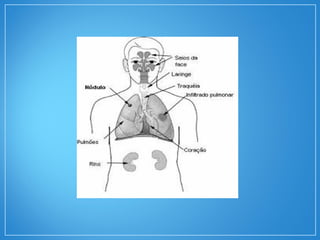
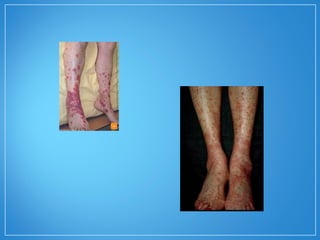
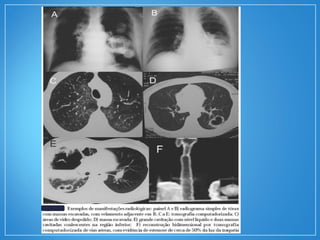
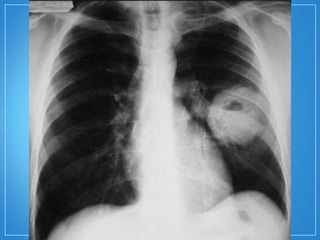
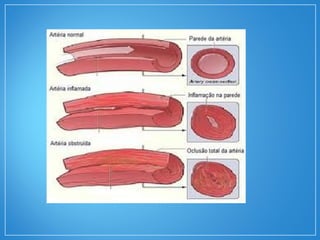
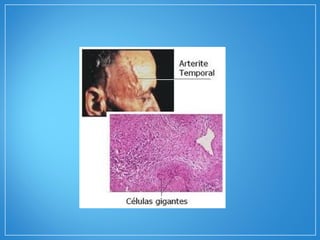
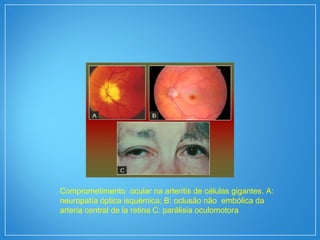
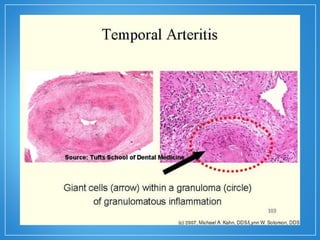
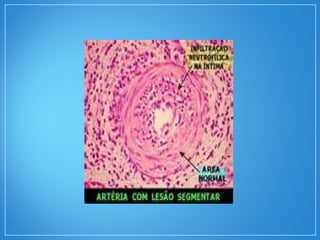
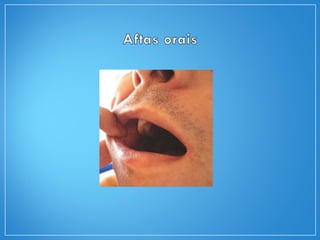
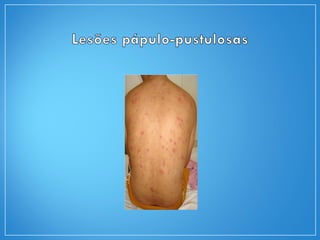
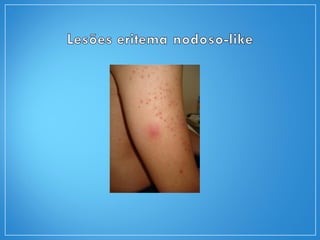
2) A granulomatose de Wegener acomete preferencialmente as vias aéreas e rins e causa tosse, hemoptise e glomerulonefrite. Já a arterite de células gigantes lesiona artérias de médio e grande porte e provoca forte cefaleia.
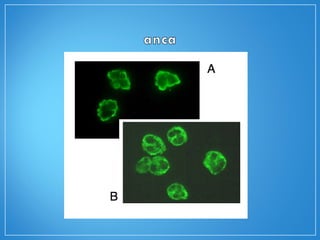
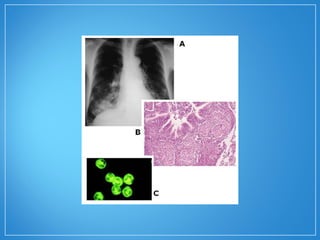
3) Os exames mostram alterações inflamatóri