Baixado 75 vezes






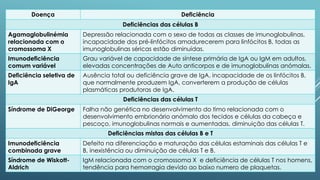
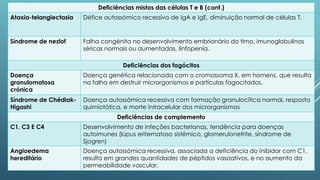




















O documento discute imunodeficiências primárias e secundárias, incluindo suas causas, sintomas, diagnóstico e tratamento. As imunodeficiências primárias resultam de defeitos genéticos nas células do sistema imunitário, enquanto as secundárias são adquiridas por fatores como infecções ou tratamentos médicos. O diagnóstico envolve testes de sangue e genéticos, e o tratamento inclui antibióticos, terapia de substituição e transplante de medula óssea em alguns casos.
















































































