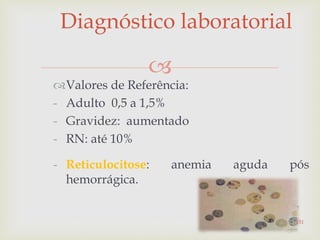
Baixado 309 vezes




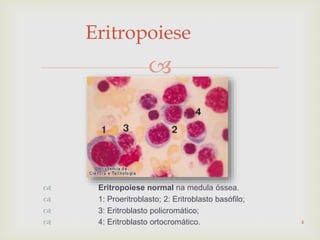
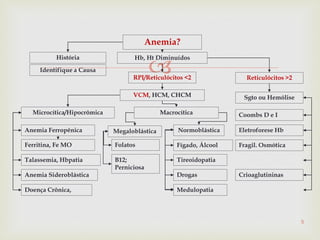






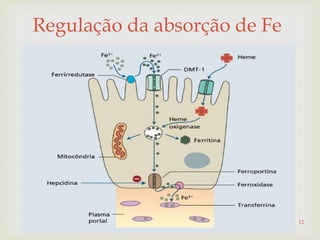



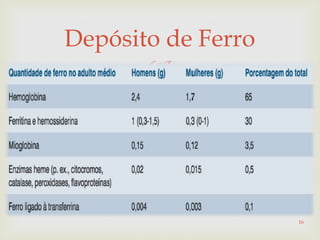








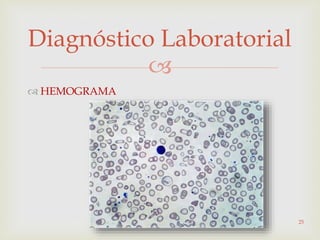




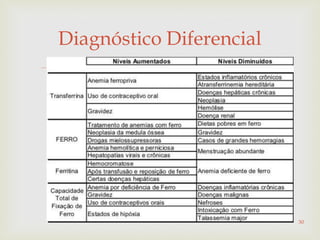

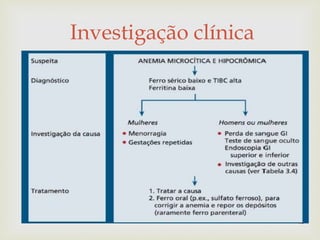



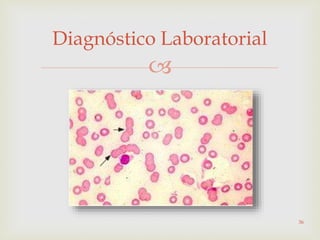




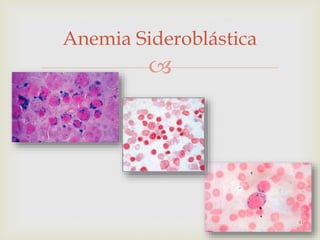

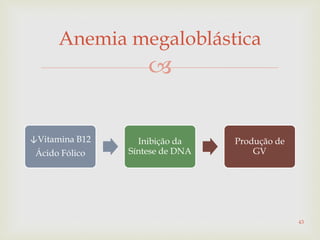







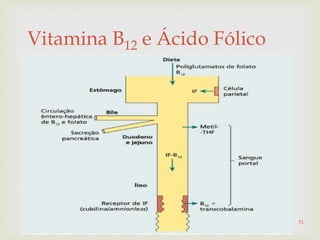
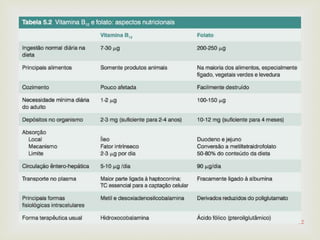
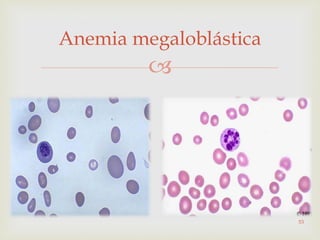
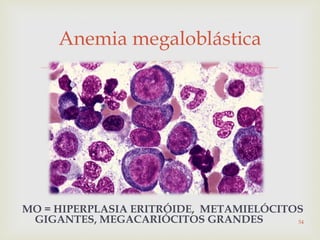

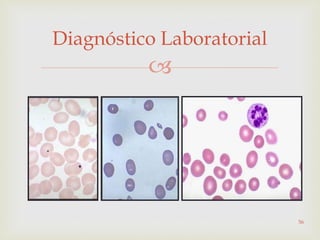
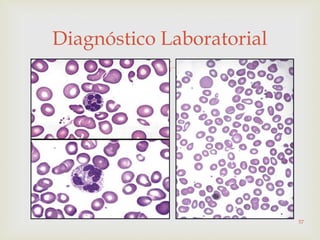















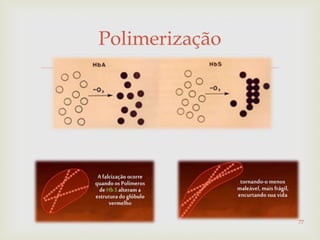

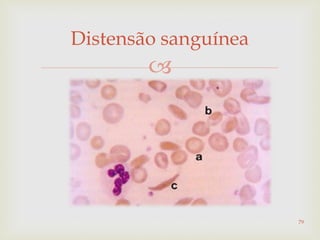



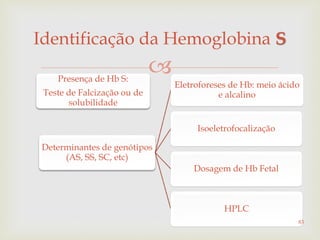
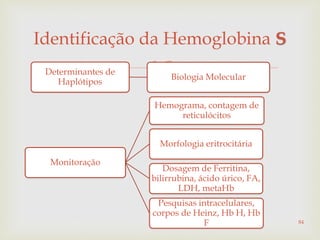


























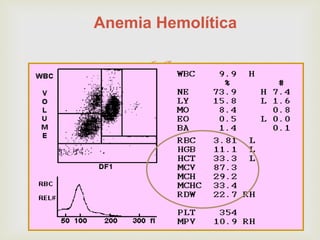

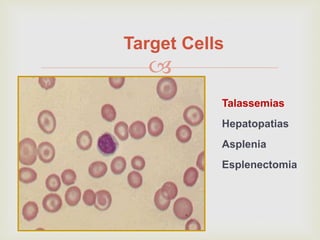
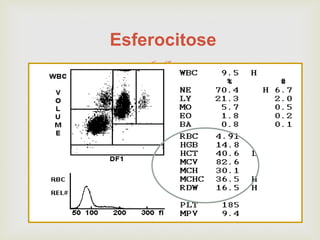
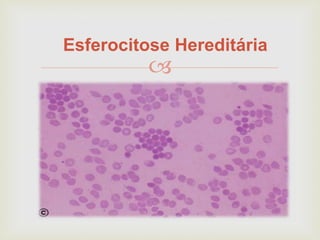




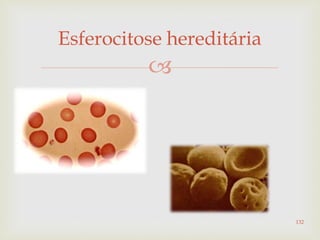







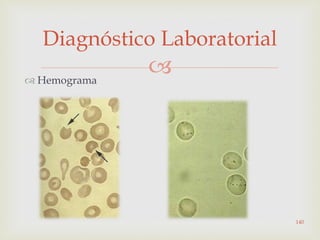




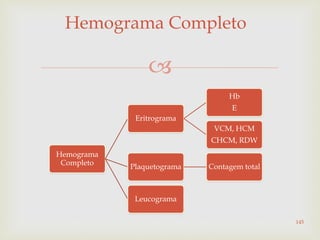


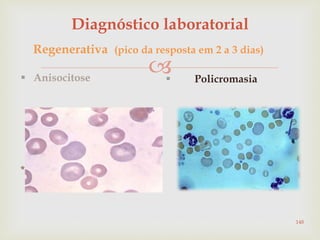



Este documento discute vários tipos de anemias, incluindo suas causas, sintomas e abordagens de diagnóstico. Ele fornece detalhes sobre a eritropoese normal e anormal, anemias microcíticas e hipocrômicas como a anemia ferropênica, anemias macrocíticas como a anemia megaloblástica e a anemia perniciosa.





























































![Anemia%20 reduzida%20[modo%20de%20compatibilidade]](https://cdn.slidesharecdn.com/ss_thumbnails/anemia20reduzida20modo20de20compatibilidade-130519081516-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)









