O documento resume os principais pontos da interpretação do hemograma, incluindo a avaliação dos elementos figurados do sangue, índices hematimétricos e classificação de anemias como ferropriva e megaloblástica.
INTERPRETAÇÃO DO HEMOGRAMA
HEMOGRAMACOMPLETO
– Exame simples e baixo custo
– Auxilia no diagnóstico de patologias hematológicas e sistêmicas
• Avaliação quantitativa e qualitatita dos elementos do sangue
• Subdividido em eritrograma, leucograma ou plaquetograma
• Contagem de eritrócitos – expressa número de hemácias por mm3
• Distensão sanguinea – visualiza as 3 series
2.
INTERPRETAÇÃO DO HEMOGRAMA
•CHCM
– Detecção de desidratação celular
– Aumentado nas doenças que afetam membrana eritrocitária como
microesferocitose e anemia hemolítica auto imune
– Na doença falciforme ocorre desidratação celular aumentando o CHCM
• RDW
– Varia 11,5 – 14,5%,
– Medida de intensidade de anisocitose
• HEMATÓCRITO( % )
_ Porcentagem da massa eritrocitárias em relação ao volume total
• HCM
_ Representa o peso da Hemoglobina média dos eritrócitos ( picogramas)
NÍVEIS DE HEMOGLOBINAINDICATIVOS
DE ANEMIA AO NÍVEL DO MAR
Grupos por faixa etária/sexo Hemoglobina g/dlGrupos por faixa etária/sexo Hemoglobina g/dl
6 meses - 5 anos < 116 meses - 5 anos < 11
6anos a 14 anos < 126anos a 14 anos < 12
Homens adultos < 13Homens adultos < 13
Mulheres adultas < 12Mulheres adultas < 12
Mulheres grávidas < 11Mulheres grávidas < 11
5.
ANEMIAS
Conceito: Diminuição damassa eritrocitária
Melhor Método para Avaliar a Massa Eritrocitária:
Dosagem de Hemoglobina
Por quê o Hematócrito não é adequado?
Intrinsecamente susceptível a variações de volume
plasmático
6.
CLASSIFICAÇÃO
Por perda sanguínea– Anemias Hemorrágicas
agudas ou crônicas
Por aumento na destruição dos eritrócitos _
HEMÓLISES (Crônicas ou adquiridas ).
Por produção inadequada de eritrócitos _
(alteração da hematopoese) carência ou
hipoplasia medular
7.
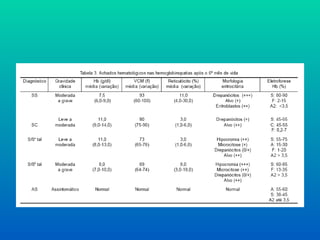
CLASSIFICAÇÃO MORFOLÓGICA
VCM= Hto(%) x 10 (expressa em
fentolitros)
GV (em milhões por mm³)
Valores de VCM:
• Microcíticas: VCM<80 fl
• Normocíticas: VCM 80-100 fl
• Macrocíticas: VCM> 100 fl
8.
Classificação Morfológica
MICROCÍTICAS:
- Deficiênciade ferro
- Intoxicação por chumbo
- Talassemias
- Anemia de origem infecciosa
NORMOCÍTICAS:
- Anemias hemolíticas
- Secundária a perdas
- Seqüestração esplênica
- Doenças malignas
- MACROCÍTICAS:
- Deficiência de vitamina B12/ ác. fólico
- Anemia aplástica
- Síndrome de Blackfan-Diamond
- Hipotireoidismo
- Hepatopatia/Nefropatia crônicas
9.
DEFICIÊNCIA DE PRODUÇÃODE
ERITRÓCITOS
• Falta de tecido eritropoiético:
– Destruição
– Estimulantes (EPO)↓
– congênita.
• Invasão da medula óssea por elementos
malignos.
• ↓ Elementos essenciais à eritropoiese:
– Ferro
– Folato
– Vitaminas B 12, B 6 e C.
10.
EXCESSO DE DESTRUIÇÃODE
ERITRÓCITOS (HEMÓLISE)
• Agressão ao eritrócito:
– Toxinas
– Parasitas
– Imunológica
• Defeito do eritrócito:
– Arquitetura da membrana
– Enzimático
– Hemoglobina anormal
• Hiperesplenismo
DIAGNÓSTICO CLÍNICO
As manifestaçõesclínicas são decorrentes da redução da capacidade
transporte de O₂ no sangue e menor oxigenação dos tecidos
(hipóxia):
• Palidez
• Tontura
• Astenia
• Falta de interesse e atenção
• Perversão do apetite ( picacismo) e anorexia
• Diminuição da libido
• Irritabilidade
• Cefaléia
• Debilidade física
• Dispnéia aos esforços
• Depressão imunológica
DIAGNÓSTICO LABORATORIAL
• Contagemde reticulócitos:
– 0,5 a 2,0%, em nº absolutos 25.000 a 75.000/ µl.
<0,5% = insuficiente produção medular.
>2,0% = hiperprodução medular.
• Mielograma:
– Normoblástica: Fe, hemólise, infecção.
– Hiperplasia da série vermelha: ↓ Fe e hemólise.
– Hipoplasia.
– Eritropoiese megaloblástica: ↓ folato e vitamina B 12.
– Invasão de células anormais
15.
ANEMIA FERROPRIVA
É adoença mais freqüente do mundo
(40 a 50% dos < 5 a)
Acomete mais de 600 milhões de
pessoas
Carência de ferro = 3 vezes mais
freqüente
16.
GRUPOS DE RISCOPARA
DEFICIÊNCIA DE FERRO
Crianças entre 6 meses e 2 - 3 anos
Mulheres em idade reprodutiva
Mulheres grávidas
Doadores regulares de sangue
Comunidades com baixa ingestão de
ferro heme
17.
ANEMIA FERROPRIVA
- Deficiênciasna ingesta de Ferro
- Deficiência na absorção de Ferro
- Aumento das necessidades:
- crescimento
- gravidez/ lactação
- menstruação
- Por perda crônica de sangue
- parasitoses
- hemorróidas
- distúrbios menstruais
- epistaxes
- úlceras
• Instalação gradual(estágio ↓ Fe corporal)
• Sintomatologia característica:
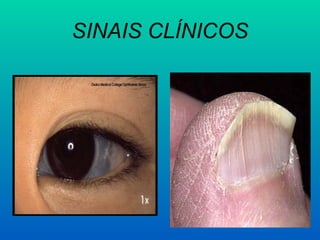
– Sintomas gerais da anemia
– Glossite atrófica
– Perversão alimentar
– Disfagia intensa
– Amenorréia
– Diminuição da libido
– Queda do rendimento intelectual
20.
DIAGNÓSTICO LABORATORIAL
• Hemograma:
–↓ Hb, Ht, VCM, HCM, CHCM
• Esfregaço : microcitose e hipocromia,
pode haver poiquilocitose e hemácias em
alvo.
• Ferro Sérico ↓ : < 70µg/dl (VR:115 ± 50)
• Transferina ↑ : > 400µg/ml
• Saturação da transferrina ↓ : < 15%
• Ferritina sérica ↓ : <10ng/ml
21.
CINÉTICA DO FERRO
•Ferro sérico
– Interpretar junto com ferritina e TIBC
– Baixo:
- anemia ferropriva (<30μg/dL)
- anemia de doença crônica (geralmente <50μg/dL)
– Normal ou elevado: talassemia e anemia sideroblástica
• Ferritina sérica
– 1º parâmetro a se alterar
– Correlacionam-se bem com os estoques de ferro corporal
– Ferropriva: <15ng/mL
– Anemia de doença crônica: 30-200ng/mL
– Talassemias e anemia sideroblástica: 50-300ng/mL
22.
CINÉTICA DO FERRO
•TIBC
– Reflete quantidade de transferrina disponível no soro para
combinar-se com ferro
– Deficiência de ferro→ TIBC >360μg/dL, por aumento da
produção de transferrina pelo fígado, em resposta à
carência de ferro
– Anemia de doença crônica → TIBC<300μg/mL)
– Normal ou elevado: talassemia e anemia sideroblástica
• Saturação da transferrina (Fe/TIBC)
– Normal: 20-40%
– Anemia ferropriva: <15%
– Anemia de doença crônica: 10-20%
24.
• Reticulócitos: normaisou levemente
elevados
• Mielograma: hiperplasia eritróide; ↓ grânulos
de Fe nos normoblastos; quase ausência de
Fe corável ou ferritina.
• Prova terapêutica: pode substituir
comprovação laboratorial.
– Administração oral de Fe, com aumento
significativo dos reticulócitos em 1 semana
e da Hb em 1 mês.
25.
TRATAMENTO DA FERROPENIA
•Correção da causa de espoliação do Fe.
• Adequação dietética.
• Sais de Fe via oral :
– 5 a 6 mg de Ferro elementar/Kg/dia
(dividido em 2 a 3 tomadas, 1 hora antes das
refeições, de preferencia com suco ácido)
Efeitos Adversos no TGI: náuseas, vômitos, desconforto
epigástrico e diarréia, nesses casos observa-se melhor
tolerância quando administrados junto as refeições.
26.
TRATAMENTO
• Falha Terapêutica
1.Diagnósticoetiológico errado.
2.Anemia é multifatorial
3.Não adesão ao tratamento
4.Ritmo do sangramento crônico maior que a
reposição
5.Doença Celíaca – Fe oral não é absorvido
27.
Indicações de FerroParenteral
1. Síndrome de Má absorção duodeno-jejunais
2. Intolerância das preparações orais.
3. Necessidade de reposição imediata dos
estoques de Fe
4. Perda de Fe excedendo a reposição oral
maxima.
28.
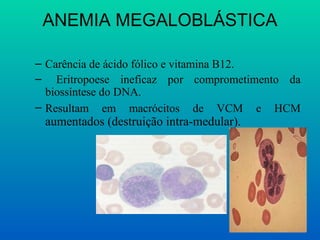
ANEMIA MEGALOBLÁSTICA
– Carênciade ácido fólico e vitamina B12.
– Eritropoese ineficaz por comprometimento da
biossíntese do DNA.
– Resultam em macrócitos de VCM e HCM
aumentados (destruição intra-medular).
29.
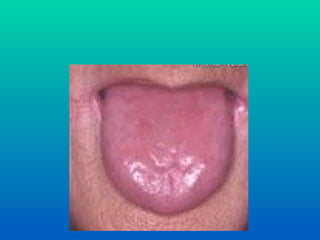
ANEMIA PERNICIOSA
Gastrite atróficaimunológica
Risco aumentado para neo gástrico
• glossite
• dormências e falta de sensibilidade nas
extremidades
• deterioração mental irreversível ( PSICOSE,
DEMENCIA )
30.
DEFICIÊNCIA DE VITAMINAB12
Vitamina B12- cianocobalamina
• Fonte: Alimentos de origem animal
• Ingestão: 5 a 15 mcg/dia na dieta ocidental,
• Necessidade diária: 2mcg.
• Estoque: 2 a 5 mg
• Tempo de instalação: 2 a 5 anos
• Digestão: Duodenal, após ligação com fator intrínseco
secretado pelo estômago
31.
DEFICIÊNCIA DE VITAMINAB12
Co-enzima de duas reações:
• Eritropoese normal (síntese de DNA),
• Conversão de metilmalonil em succinil Co-A –
acúmulo de metilmalonil, responsável pela
neuropatia.
32.
ANEMIA MEGALOBLÁSTICA
• Deficiênciade B12
1) Ingesta inadequada
2) Má absorção
- ↓Liberação do alimento (acloridria, gastrectomia parcial)
- ↓ Prod. FI (anemia perniciosa, gastrectomia total, congênita)
- Desordens do Íleo Terminal (ressecção intestinal, Chron…)
- Insuficiência pancreática
- Competição pela Cobalamina (hiperproliferação bacteriana,
Diphyllobotrium latum)
- Drogas (colchicina, neomicina…)
3) Outras (↓ Transcobalamina II, defeitos enzimáticos…)
33.
CARÊNCIA DE ÁCIDOFÓLICO
• Diminuição da síntese de purinas e timidina
• Diminuição da síntese de DNA nas células
precursoras eritropoéticas,
• Acúmulo de metabólitos tóxicos,
• Bloqueio de reações de metilação envolvidas
na expressão gênica
35.
CARÊNCIA DE ÁCIDOFÓLICO
• Folato : anel de pterina ligado ao PABA
• Fontes: vegetais verdes e frutas cítricas
• Dieta: 300 mcg/dia
• Depósito: 5 a 10 mg
• Tempo de instalação: 6 meses
• Absorção: jejuno proximal
36.
ANEMIA MEGALOBLÁSTICA
Deficiência deácido fólico
• Deficiência de folato
– 1) Ingesta inadequada (alcoólatras, adolescentes…)
– 2) Aumento das necessidades
– - Gravidez, hemólise, malignidade, hemodiálise…
– 3) Má absorção por:
– - Distúrbio primário da mucosa duodenojejunal (ex: doença celíaca…)
- Drogas: anticonvulsivantes, barbitúricos
4) Prejuízo no Metabolismo:
- Álcool
- Drogas (metotrexato, trimetoprim…)
37.
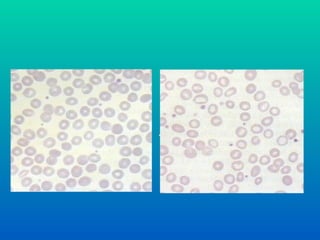
ANEMIA MEGALOBLÁSTICA
• Clínica: típica da anemia
• Hemograma: pancitopenia
• Esfregaço do sangue periférico:
neutrófilos com núcleos hipersegmentados,
hemácias com volume aumentado
38.
TRATAMENTO
Deficiência de cobalamina
100-1000µg via intramuscular por dia durante 7
dias, seguida da mesma dose 1-2 x semana, por 2
meses.
Se a causa da deficiência for anemia perniciosa ou
causa irreversível, mantem dose mensal por toda
a vida.
Reposição oral com 2 mg de vitamina B12 por dia
39.
TRATAMENTO
Deficiência de folato
1-5mg/dia via oral
Se problema de absorção, doses de até 15
mg/dia
Pacientes com necessidades continuamente
aumentadas (anemia hemolítica, má
absorção):
Ác. Fólico oral indefinidamente, com dieta rica
em folato
40.
ANEMIA ASSOCIADA AINSUFICIÊNCIA RENAL
CRÔNICA
• Anemia progressiva, conforme lesão dos
néfrons
• Multifatorial:
• Deficiencia de eritropoetina
• Diminuição sobrevida de hemácias
• Ambiente uremico
• Doença crônica/inflamação
41.
ANEMIA ASSOCIADA A
INSUFICIÊNCIARENAL CRÔNICA
• EritropoetinaEritropoetina
• indicaçõesindicações
Estimula as cels
progenitoras eritróides
imaturas
- anemia - insuf. renal crônica
- pacientes AIDS (zidovudina)
níveis endógenos de
eritropoetina
- não se destina a paciente
que necessitam de correção
imediata de anemia grave
42.
ADC
• Anemia levea moderada
• Presente em :
• Infecções,
• Inflamações
• Traumas
• Neoplasias
• Ausencia de sangramento, hemólise eou
infiltraçao tumoral
43.
ADC
• imunoinduzida?
• RETENÇÃODE FERRO NOS MACROFAGOS E
COM ISSO HIPOFERREMIA COM DIFICULDADE
NO TRANSPORTE
• BAIXA SECREÇÃO DE ERITROPOETINA NA ADC
• SOBREVIDA REDUZIDA DAS HEMÁCIAS
44.
ADC
• QUADRO CLINICODEPENDE DA DOENÇA DE
BASE
• ANEMIA LEVE, NORMO NORMO OU
LEVEMENTE HIPO-MICRO
45.
Anemias hemolíticas
• Hemóliseé definida como a destruição
prematura das hemácias na periferia, seja no
espaço intravascular, seja no interstício dos
órgãos do sistema retículo-endotelial,
provocando uma queda importante de sua
meia-vida.
46.
Classificação
das Anemias Hemolíticas
HereditáriasX Adquiridas
defeitos congênitos do eritrócito anticorpos, toxinas ou anormali
(membrana, enzima ou hemoglobina). dades da circulação.
Intracorpusculares X Extracorpusculares
anormalidades no interior dos eritrócitos - fatores extrínsecos – trauma
hemoglobinopatia ou enzimopatia mecânicos ou auto-Ac.
Intravasculares X Extravasculares
as hemácias são destruídas na hemácias destruídas no tecido
própria circulação retículo-endotelial
47.
Achados Clínicos
Icterícia leveassociada a palidez.
Esplenomegalia – talassemias.
História familiar positiva de anemia –
esferocitose hereditária, anemia
falciforme, talassemias.
Uso de medicamentos – alfa-metildopa
, dapsona.
Urina avermelhada ou marrom
(hemoglobinúria) – hemólise
intravascular.
Obs: Uma crise hemolítico aguda
intravascular freqüentemente se
manifesta com febre, lombalgia,
palidez, icterícia e “urina escura”
48.
Laboratório
• LDH elevada:285 – 1160 U/mL (LDH-2).
• Haptoglobina sérica diminuída.
• Indicadores de hemólise intravascular:
hemoglobinemia, hemossiderinúria, hemoglobinúria,
hemopexina e metemalbumina.
• Índices hematimétricos: Anemia normocítica e
normocrômica em geral. Quando a hemólise é aguda e
grave, pode aparecer macrocitose.
• Hematoscopia do sangue periférico
Anemia hemolítica Auto-imune
•Induzida pela ligação de anticorpos e/ou
complemento à membrana das hemácias.
• Geralmente causada por auto-anticorpos.
Reações transfusionais também podem ocorrer.
• Opsonização → fagocitose por macrófagos
(hemólise extravascular).
• AHAI por anticorpos quentes – IgG.
• AHAI por anticorpos frios – IgM.
51.
AHAI por IgG
•Mais prevalente.
• IgG produzida geralmente contra antígenos do sistema Rh. Principal local de
hemólise é o baço.
• 50% tem causa idiopática – mais comum em mulheres 50-60 anos.
• Alfa-metildopa, LES, LLC, Linfoma não-Hodgkin, MM, BK, CMV.
• Leve icterícia, esplenomegalia, reticulocitose 10-30%, VCM normal ou aumentado,
microesferócitos.
• Teste do Coombs Direto – positivo em 98% dos casos.
• Tratamento:
– Prednisona 1-2mg/kg/dia ou 40mg/m2
de superfície corporal.
– Esplenectomia
– Ciclofosfamida ou azatioprina
– Tto de suporte para doença crônica: ácido fólico
52.
AHAI por IgM
•Anticorpos dirigidos contra sistema (I,i). Apresentam maior atividade em
temperaturas entre 0 – 10 o
C – Crioaglutininas.
• Destruição das hemácias ocorre nos fígado pelas células de Kuppfer.
• Doença da Crioaglutinina – Idiopática: expansão clonal de linfócitos B e
produção excessiva de IgM monoclonal. 50 -70 anos.
• Doenças linfoproliferativas, infecção por Mycoplasma pneumoniae.
• Quadro clínico mais brando que a por igG. Auto-limitado nos casos
infecciosos.
• Teste de Coombs Direto e pesquisa dos títulos séricos de crioaglutininas.
• Tratamento: Imunossupressor – Clorambucil
Evitar exposição ao frio.
53.
Deficiência de G6PD
Muitocomum
Herança ligada ao cromossomo X, logo >♂ ♀
Estresse oxidativo (por infecção, uso de drogas, etc.)
2 formas GDA-
- negros, quadro mais brando e auto-limitado
GDMed
- população de ascendência do mediterrâneo, com
evolução mais grave.
Corpúsculos de Heinz - precipitados intracelulares de Hb desnaturada
produzidos pela desnaturação oxidativa da
globina.
Hemólise extra- e intravascular
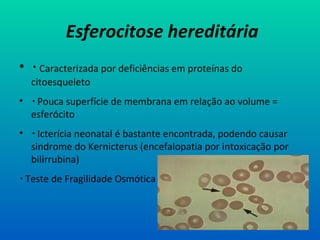
Esferocitose hereditária
• Caracterizada por deficiências em proteínas do
citoesqueleto
• Pouca superfície de membrana em relação ao volume =
esferócito
• Icterícia neonatal é bastante encontrada, podendo causar
sindrome do Kernicterus (encefalopatia por intoxicação por
bilirrubina)
Teste de Fragilidade Osmótica
56.
Esferocitose hereditária
• Doençaautossômica dominante em 75% dos
casos
• As hemácias se formam pequenas e esféricas,
não podem ser comprimidas (não possuem a
estruturuea flexível normal) leve compressão
hemólise
57.
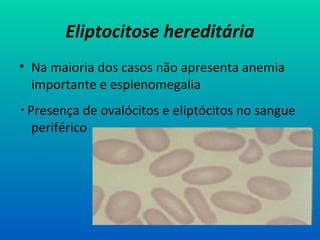
Eliptocitose hereditária
• Namaioria dos casos não apresenta anemia
importante e esplenomegalia
Presença de ovalócitos e eliptócitos no sangue
periférico
Hiperesplenismo
• Critériospara diagnóstico de hiperesplenismo:
• Citopenia de uma ou mais linhagens
hematológicas (plaquetas> hemácias >leucócitos)
• Hiperplasia reativa compensatória da medula –
a anemia tende a ser leve.
• Esplenomegalia – esplenomegalia congestiva –
cirrose hepática e esquistossomose hepatoesplênica.
60.
Anemia Hemolítica comAcantócitos
• 5% dos pacientes com cirrose hepática
avançada.
• Presença de hemácias em alvo e acantócitos.
61.
Hemoglobinúria Paroxística Noturna
(HPN)
• Desordem clonal de células tronco que originam
linhagens sanguíneas hipersensíveis ao sistema
complemento.
• Anemia aplásica abre quadro clínico em 30%
dos pacientes com HPN precipitante ? ?
• Hipercoagulabilidade 30-40% doa pacientes
apresentam eventos trombóticos.
• (Síndrome de Budd Chiari – mais clássico; e trombose
venosa cerebral)
62.
Hemólise Mecânica com
Fragmentaçãode Hemácias
• Esquizócitos – fragmentos de hemácias
devido a trauma físico excessivo do sistema
cardiovascular.
• Hemólise Traumática Macrovascular –
qualquer lesão intracardíaca que leve a
alteração da hemodinâmica, como próteses
valvares, principalmente aórtica, e estenose
aórtica calcificada grave.
63.
Hemólise traumática microvascular– associada a
doença de pequenos vasos, como PTT, síndrome
hemolítico-urêmica, CIVD, etc. deposição de
fibrina decorrente de lesão endotelial, o que leva
a fragmentação eritrocitária. Há também
trombocitopenia devido à formação de
microtrombos.
Impacto externo – hemólise do corredor
Efeitos tóxicos – queimaduras, exposição a altas
temperaturas, lise osmótica, agentes químicos,
etc.
64.
Consequências da Hemólise
•Resposta medular
– Anemia hiperproliferativa ( reticulocitose )
– Hiperplasia eritróide com aumento da produção
de reticulócitos
• Aumento da produção de bilirrubina indireta
• Formação de cálculos biliares
Obs. Anemia pós-hemorrágica aguda
também cursa com reticulocitose.
65.
Tipos Normais deHemoglobina
• HbA1: hemoglobina adulta; 2 cadeias alfa e 2 beta;= 95% do
total de Hb
• HbA2: 2 cadeias alfa e 2 delta; = 2% do total de Hb
• HbF: 2 cadeias alfa e 2 gama principal Hb no feto e recém-
nascido; 60%da Hb total até o 8º mês de gestação; substituída
porHb A1; menos de 5% no adulto
66.
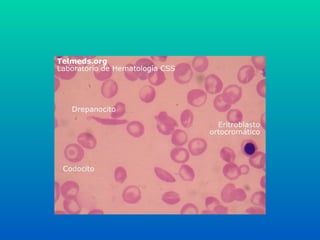
Anemia Falciforme
• Trocade um aa polar (GLU)
por um apolar (VAL) na cadeia β.
• Hb S
• A hemoglobina em baixa quantidade de
oxigênio se precipita dentro da hemácia
• FORMA DE FOICE
67.
Anemia Falciforme
• Essaforma é menos solúvel e danifica a membrana
• celular célula mais frágil hemólise
• • Ocorre principalmente na população negra (0,3-1%)
• • Doença homozigota recessiva
• • Durante toda a vida, crises de dor e susceptibilidade
• aumentada para infecções
• • reduzida tendência a contrair Malária
• • 1. Crise de Dor
• • 2. Icterícia
• • 3. Síndrome Mão-pé
• • 4. Infecções
• • 5. Úlcera de Perna
- Crise Óssea– Isquemia aguda ou infarto da medula óssea.
Acomete mais ossos longos das extremidades, coluna
vertebral e arcos costais. Dor de grande intensidade, sem
edema, calor ou rubor. Pode haver dor peri-articular de
grandes articulações periféricas. Radiografia normal.
- Crise Abdominal – Dor abdominal difusa súbita, associada à
distensão e sinais de irritação peritonial. Ocorre por
isquemia ou micro-infartos mesentéricos.
- Crise Hepática – Obstrução dos sinusóides hepáticos,
levando à hepatite isquêmica aguda. Cursa com dor local,
hepatomegalia e elevação significativa das transaminases e
bilirrubina.
70.
- Síndrome TorácicaAguda – Quadro de febre
alta, taquipnéia, dor torácica, leucocitose e
infiltrado pulmonar. Pode ocorrer por:
infecção (germe mais comum é o S.
pneumoniae, mas também podem ser
encontrados germes atípicos como
Mycoplasma pneumoniae e Chlamydia
pneumoniae, infarto pulmonar, embolia
gordurosa, TEP e infarto costal.
71.
priapismo
-Priapismo – Afoiçamento
dehemácias obstruem os
sinusóides do corpo
cavernoso. Mais comum nas
faixas etárias de 5-13 anos
(priapismo puberal) e de 21-
29 anos (priapismo pós-
puberal). Definida como
ereção prolongada e
dolorosa.
72.
• Infecções
- Principalcausa de mortalidade.
- Predisposição devido à ↓ função esplênica, ↓ da
capacidade de fagocitose e defeito na ativação da via
alternativa do complemento.
- Bacteremia = S. pneumoniae, H. influenzae b, Gram-
negativos → 20-50% mortalidade
- Meningite = S. pneumoniae, H. influenzae b
- Pneumonia = infecção mais comum. Mycoplasma
pneumoniae, vírus respiratórios, S. pneumoniae.
- Osteomielite = Salmonella, Staphylococcus aureus.
73.
• Crise deSeqüestro Esplênico
- São caracterizadas pelo aprisionamento de
eritrócitos, especialmente no baço.
- Exacerbação aguda da anemia, reticulocitose
persistente, baço hipersensível e de tamanho
aumentado e, por vezes, hipovolemia.
- Alto risco em pacientes que não tem baço
atrófico.
- Recidiva em 50% dos casos, sendo indicada
esplenectomia.
74.
• Crise Aplásica
-Consiste em parada transitória da eritropoese,
caracterizada por quedas abruptas dos níveis de
hemoglobina, contagem de reticulócitos e
precursores eritróides da medula óssea.
- Parvovírus B19 responsável por 68% das crises em
crianças. Em adultos a percentagem é menor.
- Necrose da medula óssea também provoca crise
aplásica.
75.
•Anemia Crônica
-Os eritrócitossão destruídos randomicamente, com tempo
de sobrevida médio de 17 dias.
-Além da hemólise, os níveis inapropriadamente baixos de
eritropoetina contribuem para a anemia.
•Crise Hemolítica
-Taxa aumentada de hemólise com queda da hemoglobina e
elevação da contagem de reticulócito
-São raras e muitas vezes confundidas com outras causas de
icterícia como hepatite, litíase biliar,...
-deficiência concomitante da glicose-6-fosfato desidrogenase
(?)
76.
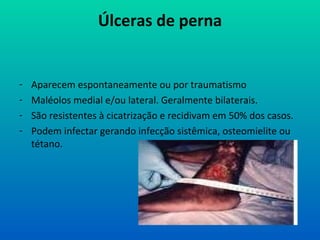
Úlceras de perna
-Aparecem espontaneamente ou por traumatismo
- Maléolos medial e/ou lateral. Geralmente bilaterais.
- São resistentes à cicatrização e recidivam em 50% dos casos.
- Podem infectar gerando infecção sistêmica, osteomielite ou
tétano.
77.
Complicações Ósseas
- Síndrome“mão-pé”: tumefação dolorosa das superfícies
dorsais de mãos e/ou pés.
- acomete metade das crianças com AF. Incidência entre 1-4
anos.
- causada pela oclusão dos capilares nos ossos pequenos dos
membros.
78.
- Osteonecrose dacabeça do fêmur
- Infarto da medula óssea: pode provocar
exacerbação da anemia, pancitopenia,
embolia gordurosa pulmonar.
- a presença de necrose na MO pode
favorecer o desenvolvimento de infecções,
especialmente por Salmonella e S. aureus
79.
Hemólise
• Predominantemente extravascular– macrófagos do
tecido reticulo-endotelial, principalmente esplênicos.
• Mecanismos:
- perda de elasticidade e deformabilidade;
- opsonização por auto-anticorpos IgG;
• As hemácias afoiçadas são as primeiras a serem
fagocitadas.
• Meia-vida média de 20 dias.
• Anemia crônica
• Icterícia
• Colelitíase
80.
Manifestações clínicas
• Apartir de 6 meses de idade
• Até esta idade, o percentual de HbF é grande!
• A HbF tem propriedades físico-químicas
diferentes da HbS, e não participa do
polímero.
• Efeito diluidor → quanto maior a
concentração de HbF, menor a polimerização
de HbS → menor afoiçamento.
Diagnóstico da AnemiaFalciforme
• Hemograma e bioquímica
• Esfregaço de sangue periférico
• Teste do afoiçamento
• Eletroforese de hemoglobina
• Hemoglobina: ↓
• Hematócrito: ↓
• Índices hematimétricos normais
• Reticulócitos: ↑
• LDH: ↑
• Haptoglobina: ↓ ou ausente
• Bilirrubina indireta: ↑
85.
Síndrome torácica aguda
•Febre, taquipnéia, dor torácica, leucocitose, infiltrado pulmonar;
• O aparecimento de alterações radiológicas pode ser tardio;
• É uma forma de doença pulmonar potencialmente muito grave;
• Na anemia falciforme, a incidência correlaciona-se com baixos
níveis de HbF e altos níveis de leucócitos. Nos adultos há
comumente crise dolorosa 2-3 dias antes do aparecimento da STA.
• Fatores preciptantes mais comuns: infecção (Chlamydia
pneumoniae e Mycoplasma pneumoniae), infarto pulmonar e
embolia gordurosa por infarto de medula óssea.
• Tem um bom prognóstico, se devidamente tratada.
86.
Tratamento
• Medidas gerais:
-Visitasclínicas de rotina;
-Aconselhamento genético;
-Ingestão hídrica adequada;
-Evitar frio ou calor excessivos;
-Evitar traumas e estresse;
-Ingestão diária de ácido fólico 1mg VO;
-Penicilina VO diária ou IM mensal até 6 anos;
-Vacinação contra pneumococo, meningococo, H.
influenzae;
87.
tratamento
• Hemotransfusão;
• Tratamentodas crises e complicações;
• Hidroxiuréia;
• Transplante de medula óssea
- reservado para crianças que apresentam
complicações severas;
- dificuldade de encontrar doador compatível
88.
• Crise álgica:hidratação vigorosa, AINE, morfina
em casos graves, transfusão em casos extremos;
• Síndrome torácica aguda: hidratação,
oxigenoterapia, hemotransfusão para manter
hematócrito > 30 %; Exosangüíneo-
Transfusão(SatO2 < 90%);
• Infecção: cefalosporina de 3ª geração;
• Úlcera: repouso, cuidados locais,
antibioticoterapia sistêmica (se infectada). Após
debridamento das áreas necróticas utilizar óxido
de zinco local.
89.
Indicações para hemotransfusão
Definitivas:
•Evento neurológico agudo
• Seqüestro esplênico
• Pneumonia grave ou infarto pulmonar
• Anemia grave com descompensação cardíaca
• Anemia aplásica
• Pré-operatório
90.
• Relativas:
• Anemiasintomática
• Seqüestro hepático
• Úlceras de MI refratárias ao tratamento
• Priapismo refratário ao tratamento
• Episódios álgicos graves, prolongados ou
freqüentes
• Insuficiência respiratória crônica
• Gravidez
91.
Hidroxiuréia
• É umadroga utilizada na terapia do câncer;
• Age aumento da concentração de HbF, diminuindo a
polimerização da HBS e, conseqüentemente, o
afoiçamento.
• Efeitos adversos: leucopenia e aumento do VCM pela
megaloblastose.
• Episódios álgicos freqüentes;
• História de síndrome torácica aguda;
• Eventos vaso-oclusivos diversos, principalmente SNC;
• Anemia severa.
92.
Indicações :
pelo menosuma das seguintes complicações nos últimos 12
meses:
• 3 ou mais episódios de crises vaso-oclusivas com
necessidade de atendimento médico;
• 1 crise torácica aguda recidivante (definida como dor
torácica aguda com infiltrado pulmonar novo,
• febre de 38,5 °C ou mais, taquipnéia, sibilos pulmonares
ou tosse)10;
• 1 ou mais acidentes vasculares encefálicos ou ataques
isquêmicos transitórios mesmo que em
• programas de transfusão de substituição;
• 1 episódio de priapismo grave pós-puberal ou priapismo
recorrente;
• anemia grave e persistente (Hb < 6,0 g/dl em três
dosagens no período de 3 meses).
93.
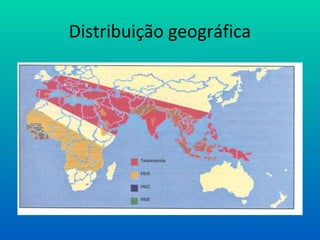
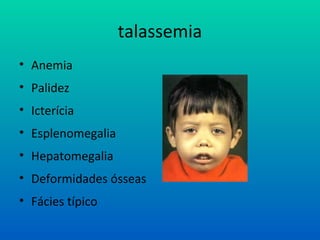
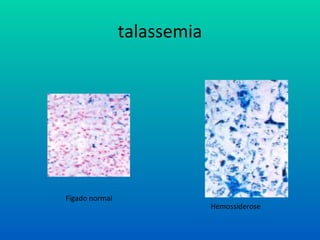
Talassemia
Síntese desequilibrada deglobina
• quantidades menores de Hb funcionante
• Talassemia α (defeito na formação das cadeias α)
• Talassemia β (defeito na formação das cadeias β)
• Traço talassêmico: Heterozigotos
• elevada prevalência nos povos do mediterrâneo, sendo a
doença genética mais comum em todo o mundo.
• Talassemias Menores (apenas um gene):discreta anemia
• Talassemias Maiores (dois genes): anemia severa, icterícia,
• deformidades ósseas e esplenomegalia
Talassemia tratamento
• Alimentaçãosaudável
• Ácido fólico
• Hemotransfusões
• Esplenectomia
• Apoio psicológico e social
• Sobrecarga de ferro
– Desferroxamina (Desferal)
– Deferiprone (Ferriprox)
• Transplante de medula óssea
102.
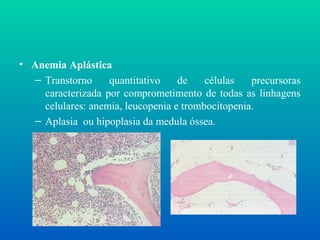
• Anemia Aplástica
–Transtorno quantitativo de células precursoras
caracterizada por comprometimento de todas as linhagens
celulares: anemia, leucopenia e trombocitopenia.
– Aplasia ou hipoplasia da medula óssea.
Aplasia de medulaossea
• É caracterizada por pancitopenia com medula
óssea hipocelular , por redução dos
precursores medulares.
• Pode ocorrer aplasia de apenas uma série
hematopoiética , principalmente nas aplasias
congênitas











































































































































![Anemia%20 reduzida%20[modo%20de%20compatibilidade]](https://cdn.slidesharecdn.com/ss_thumbnails/anemia20reduzida20modo20de20compatibilidade-130519081516-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)





