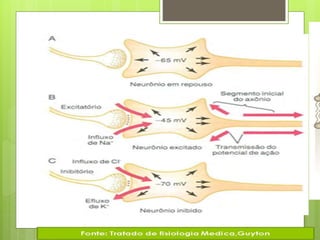
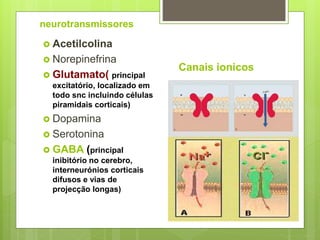
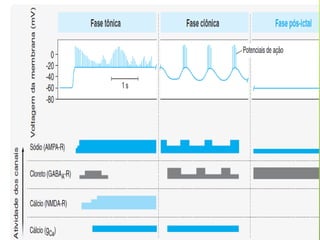
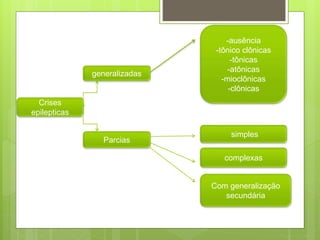
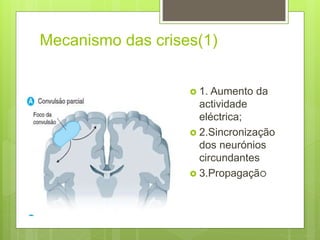
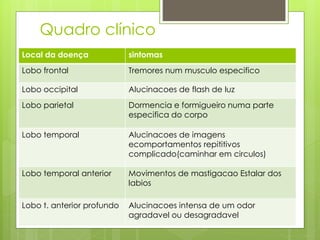
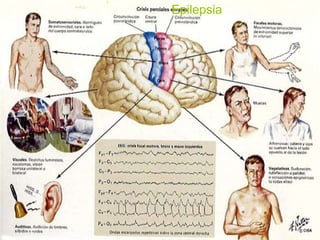
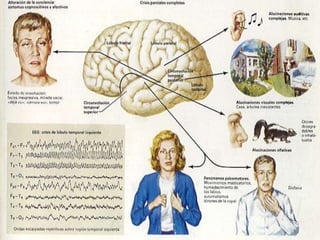
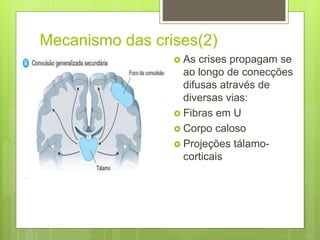
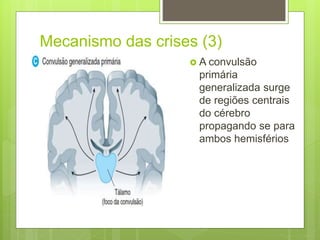
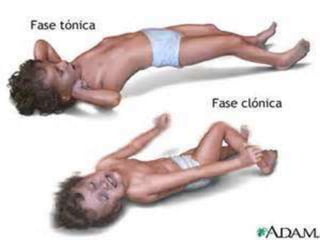
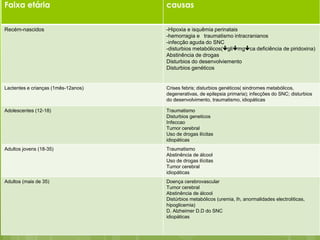
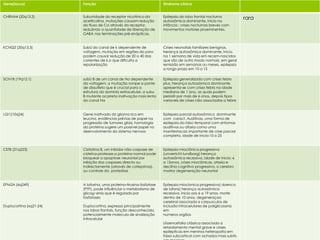
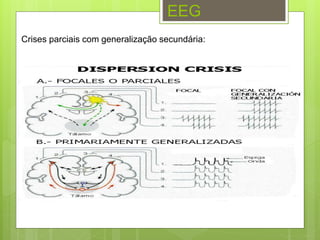
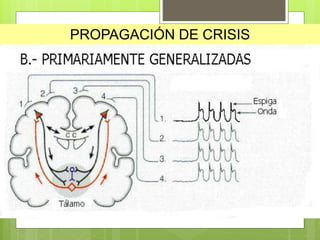
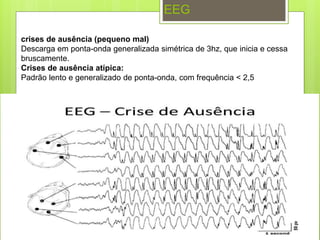
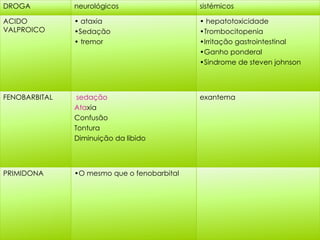
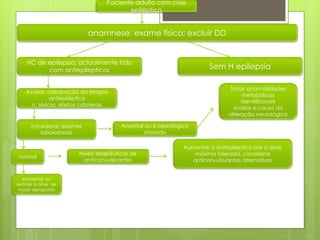
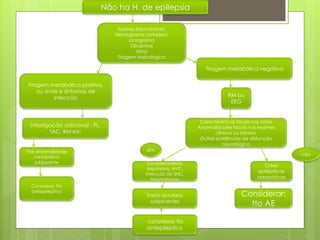
O documento discute a epilepsia, incluindo sua fisiopatologia, classificação de crises epilépticas, causas e síndromes associadas. Aborda os mecanismos das crises no nível celular e como elas se propagam no cérebro, além de tratar dos principais genes ligados a formas hereditárias da doença.