Transferir como PDF, PPTX














































![Importância do polimorfismo
• Diferentes formas polimórficas tem
diferentes propriedades quimicas e
físicas:
– Ponto de fusão, Reatividade
quimica, Solubilidade aparente,
Taxa de dissolução,
Propriedades opticas e
mecânicas, Pressão de vapor,
Densidade
• Estas propriedades tem um efeito
direto na habilidade de processar
e/ou fabricar o fármaco ou o
produto acabado:
– Estabilidade do produto
acabado, Dissolução,
Bioequivalência
61
5-Methyl-2-[(2-nitrophenyl)amino]-3-
thiophenecarbonitrile has been crystallized in ten
polymorphs: The structure of themolecule is shown in
the figure. The systemhas been named ROY for its
red,orange, andyellowcrystal colors.
Thedifferentpolymorphs arenamedas, yellowprisms (Y),
red prisms (R), orange needles (ON), orange plates
(OP), yellowneedles (YN), orange-red plates (ORP), and
red plates (RPL).](https://image.slidesharecdn.com/aula-vanessa-pderelatriodeproduo-abril20141-150218160448-conversion-gate02/85/Documentacao-tecnica-e-desenvolvimento-de-produtos-61-320.jpg)



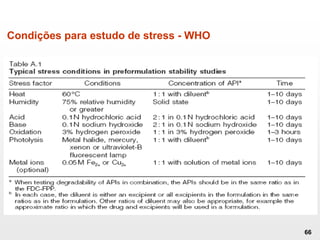







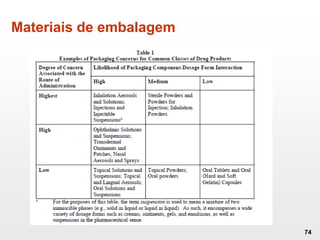



















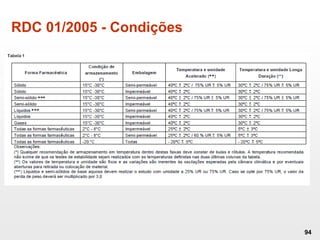

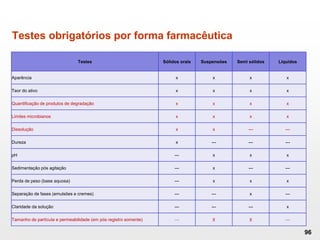

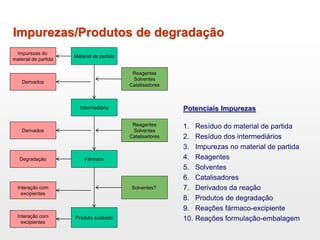




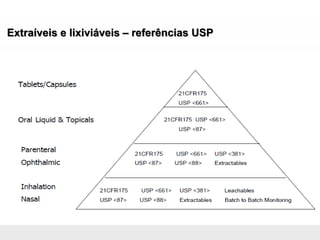



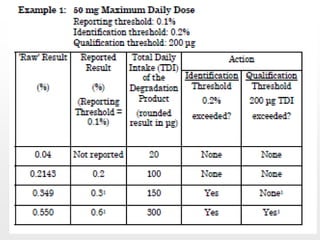


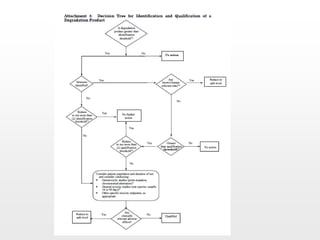







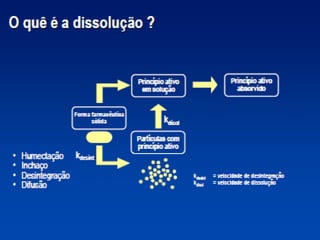


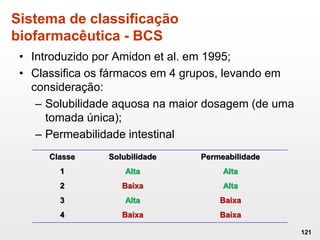












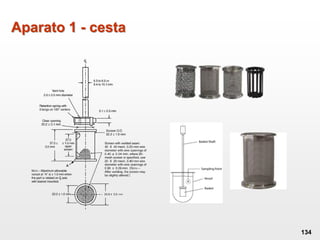
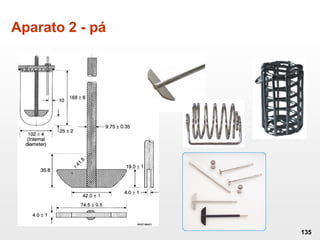










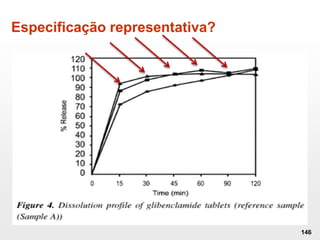




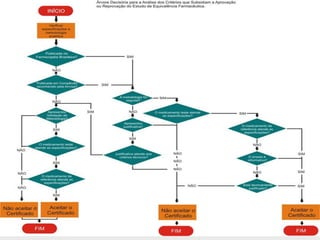




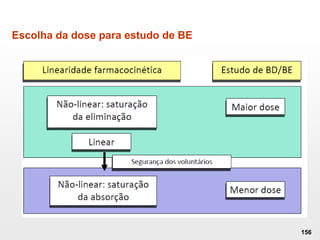
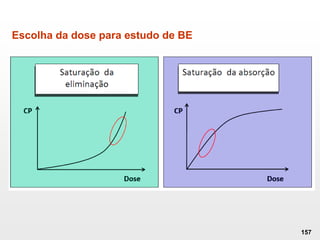
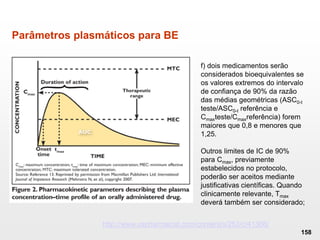

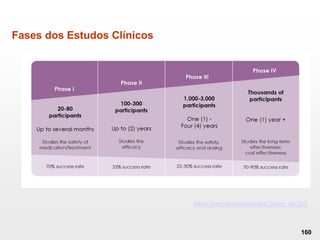

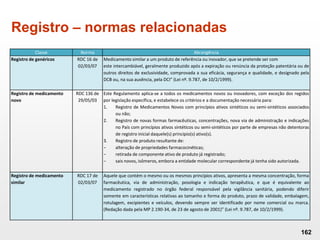
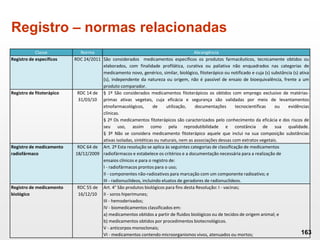




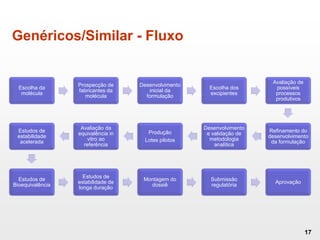
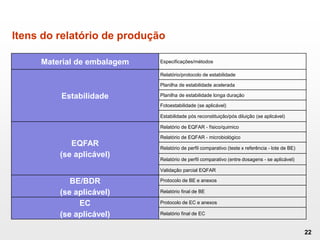
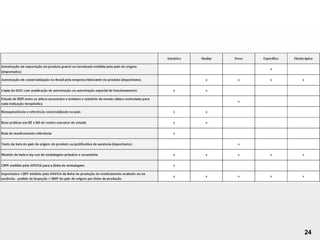
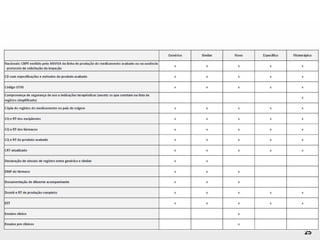
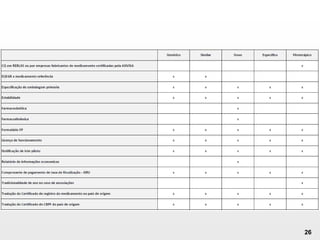
O documento discute os principais aspectos do desenvolvimento, fabricação e controle de qualidade de medicamentos, incluindo: 1) Legislações nacionais e internacionais aplicáveis às áreas de pesquisa e desenvolvimento, produção e controle de qualidade; 2) Processos de desenvolvimento de medicamentos, incluindo escolha de matérias-primas, testes de desenvolvimento e requisitos regulatórios; 3) Testes obrigatórios e especificações aplicáveis aos produtos farmacêuticos de acordo com sua forma e legislação.












































































