Baixado 161 vezes









![Introdução aos Métodos Electroanalíticos
Análise Instrumental II 5
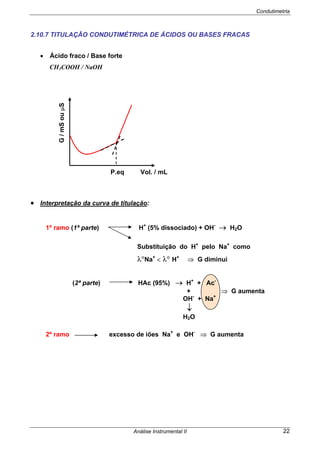
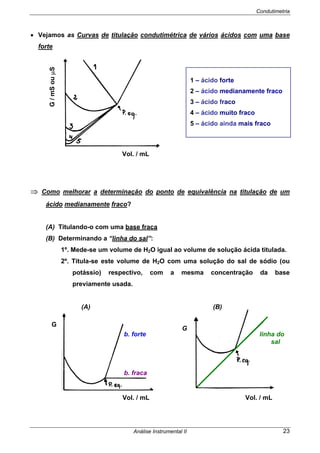
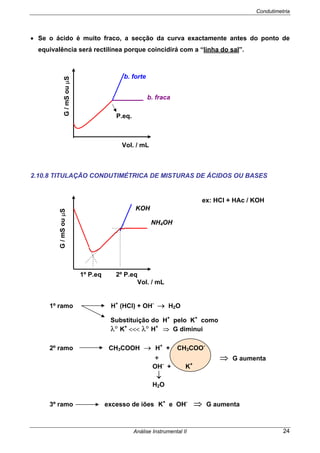
São frequentemente designados por potenciais termodinâmicos ou teóricos,
porque dizem respeito a células nas quais não passa corrente. Há factores que têm
que ser tomados em consideração quando passa corrente na célula.
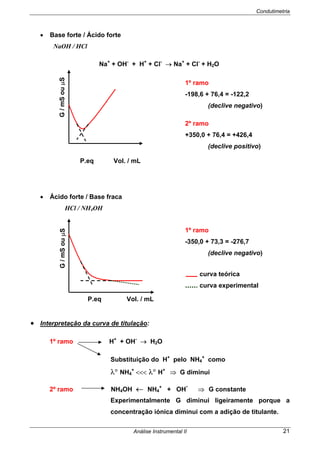
Se Ecélula> 0 as reacções são espontâneas no sentido (esq/dir)
Se Ecélula< 0 as reacções não são espontâneas no sentido indicado
⇒ A espontaneidade do processo pode ser provada recorrendo à Termodinâmica:
⇒ O Edireita e Eesquerda são os potenciais de cada semi-célula, obtidos pela
EQUAÇÃO DE NERNST.
• Para a semi-reacção de redução genérica:
Ox + ne-
↔ Red
o potencial de eléctrodo (E) é dado por:
Importante: Para a maior parte dos cálculos que envolvem baixas concentrações,
as actividades podem ser substituídas por concentrações molares.
Equação de Nernst
[ ]
[ ]ox
red
log
n
0,0592
EE 0
red
ox
−= t = 25°C (T=298,15 K)
ln = 2.303 log
(ox)
(red)
ln
nF
RT
EE 0
red
ox
−=
∆G = -nFEcel
Variação de energia livre da reacção de célula](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-10-320.jpg)














![Potenciometria
Análise Instrumental II 27
• O potencial da célula é dada por:
Edir Eesq
Eref – potencial do eléctrodo de referência; Ej – potencial de junção líquida;
Eind – potencial do eléctrodo indicador que é calculado através da EQUAÇÃO DE
NERNST.
• Para a semi-reacção de redução genérica:
aA + bB + ne
-
↔ cC + dD
o potencial de eléctrodo (E) é dado por:
E
0
Oxi/Red - Potencial Padrão de Eléctrodo, que é característica de cada semi-
reacção, V;
R - constante dos gases perfeitos, 8.314 J K-1
mol-1
T - temperatura absoluta, K
F - constante de Faraday, 96485 C mol-1
N - nº de moles de electrões que aparecem na semi-reacção para o processo
descrito
• Substituindo os valores numéricos das constantes (R e F), convertendo o
logaritmo neperiano à base 10, utilizando concentrações molares e uma
temperatura de 298,15 K:
Equação de Nernst
[ ] [ ]
[ ] [ ]ba
dc
0
red
oxind
BA
DC
log
n
0592,0
EE −=
t = 25°C (T=298,15 K)
ln = 2.303 log
( )
( )ba
dc
0
red
oxind
B(A)
D(C)
ln
nF
RT
EE −=
Ecel = Eind – Eref + Ej](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-32-320.jpg)

















![Potenciometria
Análise Instrumental II 45
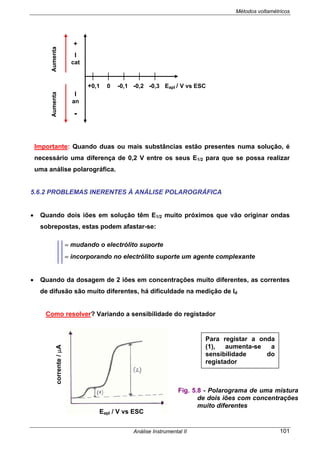
• Traça-se a curva E cel vs log [ião]
Importante:
⇒ Não se extrapolam Rectas de Calibração.
⇒ Para se aplicar o Método da Curva de Calibração, as soluções-padrão terão
que ser idênticos à amostra em termos de pH, força iónica, viscosidade, tipo
de interferentes, etc.
⇒ No caso da análise de amostras complexas isso é impossível verificar-se.
Nestes casos determina-se a concentração da amostra através do Método de
Adição de Padrão.
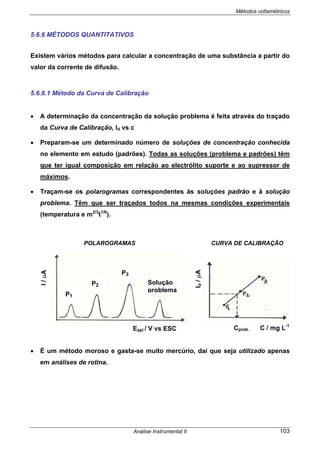
MÉTODO DE ADIÇÃO DE PADRÃO
• O potencial da célula é medido antes e depois da adição, à toma de amostra a
analisar, de vários volumes pequenos de solução-padrão.
• Para determinar a concentração de um anião em solução, a equação da resposta
do eléctrodo selectivo adequado à análise é:
ialog
nF
TR303,2
'KE −=
O potencial do eléctrodo
indicador varia linearmente com
o log [ião]
E cel
E amostra
log [ião]amost log [ião]](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-50-320.jpg)


![Potenciometria
Análise Instrumental II 51
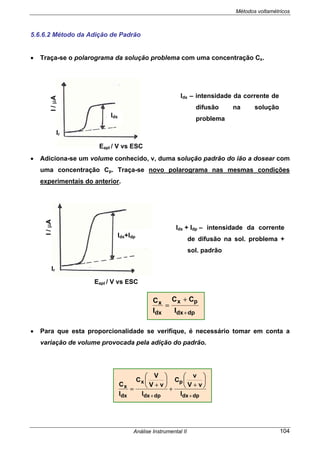
3.5.2.2 TITULAÇÃO POTENCIOMÉTRICA ÁCIDO - BASE
Nestas titulações o par de eléctrodos utilizado é o seguinte: o eléctrodo indicador
é o eléctrodo de membrana de vidro e o eléctrodo de referência pode ser ou o ESC
ou o Ag/AgCl, qualquer deles saturado em KCl. Actualmente utiliza-se o eléctrodo
combinado de pH.
Ka [HA] ≥ 10
-8
Limite para os ácidos fracos
poderem ser titulados
Importante: Não se pode fazer uma titulação potenciométrica de um ácido fraco
com uma base fraca porque o salto não é suficientemente visível. (Relembre-se que
em condutimetria isto é possível).
O salto de pH no ponto de equivalência
vai depender das forças entre ácidos e
bases.
pH
Vol. base
Vol. ácido
pH
Vol. / mL
À medida que o ácido se torna mais
fraco, o pH no ponto de equivalência
vai para valores mais altos.](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-56-320.jpg)
![Potenciometria
Análise Instrumental II 53
3.5.2.3 TITULAÇÃO ÁCIDO-BASE EM SOLVENTES ORGÂNICOS
• Muitos ácidos e bases são demasiado fracos para poderem ser titulados em meio
aquoso, mas podem ser titulados em solventes orgânicos.
• Eléctrodos eléctrodo de membrana de vidro
ESC ou Ag/AgCl, KCl (sat)
• Importante – Os eléctrodos devem ser conservados em água entre as titulações
para evitar a desidratação do vidro e a precipitação do KCl na ponte salina.
• A escala do potenciómetro a usar deve ser a de milivolts em vez da de pH, pois se
fizermos a aferição do aparelho com tampões aquosos a escala de pH não tem
significado em solventes orgânicos.
3.5.2.4 TITULAÇÃO POTENCIOMÉTRICA DE PRECIPITAÇÃO
• São muito características as reacções de precipitação dos halogenetos (Cl-
, I-
, F-
)
com o ião Ag+
.
• Eléctrodos:
⇒ eléctrodos indicadores – são os de 2ª ordem: Ag/AgCl, Ag/AgI, Ag/AgBr
⇒ eléctrodos de referência – ESC ou o Ag/AgCl saturado em nitrato de potássio.
• Exemplo: Precipitação do Cl-
/ AgNO3 com formação de AgCl
r. electródica: Ag+
+ e-
Ag
(1) [ ]++= +
Aglog0,0592EE 0
Ag
Ag
ind
Como: [ ] [ ]−
=+
Cl
AgClpsK
Ag](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-58-320.jpg)
![Potenciometria
Análise Instrumental II 54
(2) [ ]−
+
−+= Cllog0,0592psKlog0,0592EE 0
Ag
Ag
ind
(2) O indicador, traduzirá antes do ponto de equivalência a diminuição da potencial
do eléctrodo concentração do ião Cl-
.
(1) E depois do ponto de equivalência o excesso de Ag+
em solução.
3.5.2.5 TITULAÇÃO POTENCIOMÉTRICA DE UMA MISTURA DE IÕES
• É possível titular uma mistura de iões quando um dos compostos formados é
marcadamente menos solúvel.
Kps – produto de solubilidade
• Exemplo: Doseamento de uma mistura de iões I-
e Cl-
KpsAgCl = 1,82 x 10-10
KpsAgl = 8,3 x 10-17
sal mais insolúvel
• A Curva de Titulação apresenta 2 pontos de equivalência
V1< >I
-
V2< >Cl
-
6
2ps
1ps
10
K
K
≥
[ ]−−+= +
llog0,0592psAgIKlog0,0592EE 0
Ag
Ag
ind
[ ]−−+= +
Cllog0,0592AgClpsKlog0,0592EE 0
Ag
Ag
ind
⎥⎦
⎤
⎢⎣
⎡ ++= +
Aglog0,0592EE 0
Ag
Ag
ind
E / V
Vol. AgNO3 / mL](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-59-320.jpg)
![Potenciometria
Análise Instrumental II 55
3.5.2.6 TITULAÇÕES POTENCIOMÉTRICAS DE OXIDAÇÃO -REDUÇÃO
• Estas titulações envolvem a transferência de electrões da substância a ser
oxidada para a substância a ser reduzida.
• Eléctrodos:
⇒ eléctrodo indicador – eléctrodo inerte ex: platina
⇒ eléctrodo de referência – ESC ou o Ag/AgCl saturado em KCl.
• O salto de potencial verificado no ponto de equivalência depende da diferença
entre os potenciais da espécie oxidada e da espécie reduzida. Quanto maior a
diferença maior é o salto.
3.6 PROBLEMAS
1. Deduzir se uma lâmina de chumbo (E0
Pb
2+
/Pb = - 0,126 V) mergulhada numa
solução ácida de pH = 4 e [Pb2+
] = 0,100 eq L-1
, poderá servir como eléctrodo de
1ª ordem?
2. Calcule o potencial teórico da seguinte célula (considere que as actividades são
aproximadamente iguais à concentração molar e que a temperatura é de
aproximadamente 25°C):
Hg ⏐ Hg2Cl2 (sat), KCl sat⏐⏐ Fe3+
(0,84 g L-1
), Fe2+
(1,24 g L-1
) ⏐Pt
M (Fe) = 55,9 g mol-1
; E (ESC) = +0,244 V ; E0
Fe3+/Fe2+ = +0,771 V
3. Considere a seguinte reacção: CuBr (s) + e-
↔ Cu (s) + Br-
3.1 Calcule o seu potencial padrão (E0
).
3.2 Represente esquematicamente a célula que pode ser utilizada para dosear o
Br-
, em que o eléctrodo indicador de cobre é o cátodo e o ESC o ânodo.](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-60-320.jpg)
![Potenciometria
Análise Instrumental II 56
3.3 Deduza uma equação que relacione o potencial da célula representada na
alínea anterior com a [Br
-
] (considere que o Ej = 0).
3.4 Calcule o valor da [Br
-
] se o potencial da célula for de - 0,071 V.
E (ESC) = +0,244 V ; E0
Cu+/Cu = +0,518 V ; Kps(CuBr) = 5,2 x 10-9
4. A célula representada a seguir é utilizada para determinar a concentração de
CrO4
2-
:
Ag ⏐ Ag2CrO4 sat, CrO4
2-
(x mol L-1
) ⏐⏐ Hg2Cl2 (sat), KCl sat⏐ Hg
Calcule essa concentração se o potencial da célula for de - 0,402 V
5. A célula representada a seguir tem um potencial de 0,124 V:
ESC ⏐⏐ Cu2+
(a = 3,25 x 10-3
mol L-1
) ⏐ eléctrodo de membrana de Cu2+
Quando o eléctrodo de membrana de Cu2+
foi mergulhado numa solução de Cu2+
de concentração desconhecida, o potencial da célula foi de 0,105 V. Calcule a
actividade de Cu2+
nesta solução.
E (ESC) = +0,244 V
6. Na titulação potenciométrica de 10,0 mL de H3PO4 com NaOH 0,100 eq L-1
,
gastou-se até ao 2º ponto de equivalência 9,00 mL de titulante. A amostra foi
diluída até 200 mL com água destilada.
M (H3PO4) = 98 g mol-1
6.1 Determine a massa de ácido que estava dissolvida na solução.
R: 0,0441 g
6.2 Determine a concentração da solução de H3PO4 em g L-1
e em mol L-1
.
R: 4,41 g L-1
e 0,045 mol L-1](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-61-320.jpg)







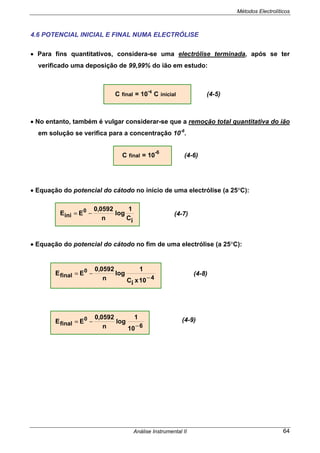



![Métodos Electrolíticos
Análise Instrumental II 69
• Exemplo – Depósito de Cobre a partir de uma solução de iões Cu2+
Cu2+
+ 2 e-
Cu E0
Cu
2+
/ Cu = +0,337 V
2 H+
+ 2 e-
H2 E0
2H
+
/ H2 = 0,000 V
NO3
-
+ 10 H+
+ 8 e-
NH4
+
+ 3 H2O E0
NO3
-
/ NH4
+
= +0,2... V
O despolarizante tem que ser escolhido com base nos potenciais normais das
espécies envolvidas.
• Uma ELECTRÓLISE (electrogravimetria) pode ser conduzida de 2 formas
diferentes:
⇒ A Intensidade de corrente constante
⇒ A Potencial de eléctrodo controlado
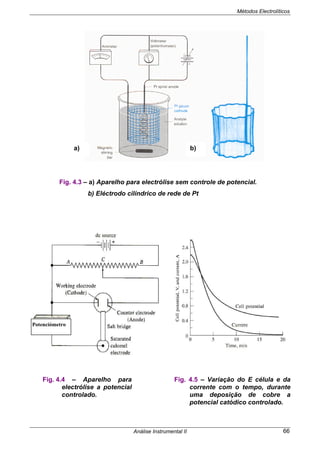
4.7.3 ELECTRÓLISE A INTENSIDADE DE CORRENTE CONSTANTE
(Fig.4.3)
• É o processo mais antigo de realizar uma electrólise.
• Impõem-se um valor de corrente por um período de tempo suficiente de modo a
dar-se a reacção completa das espécies em estudo.
• Ocorrerá em 1º lugar, no cátodo, a reacção com o potencial mais positivo e assim
sucessivamente por ordem dos potenciais catódicos ( + - ).
• Exemplo – Se através duma solução de iões Cu2+
e Zn2+
, onde estão imersos dois
eléctrodos de Pt, fizer passar uma corrente de intensidade constante, quais as
reacções catódicas que ocorrem? (Considere [Cu2+
] = [Zn2+
] = 1 mol L-1
, pH = 1
e ηH2 = - 0,29 V).
E0
Cu
2+
/ Cu = 0,337 V E0
Zn
2+
/ Zn = - 0,763 V E0
H
+
/ H2 = 0,000 V](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-74-320.jpg)




![Métodos Electrolíticos
Análise Instrumental II 74
• Aplicação deste tipo de electrogravimetria à análise de ligas
⇒ Só se analisam elementos vestigiários.
⇒ Estes elementos têm que ser mais electronegativos que a matriz da liga.
Matriz da liga - componente maioritário da liga.
4.7.6 PROBLEMAS
1. Considere que a célula representada a seguir tem uma resistência interna de
6,74 Ω e é percorrida por uma corrente de 75 mA
Pt⏐V3+
(5,1x10-4
g L-1
), V2+
(8,4 g L-1
)⏐⏐Br
-
(12,0 g L-1
), AgBr(sat)⏐Ag
Calcule o potencial esperado inicialmente para esta célula.
M (V) = 51 g mol-1
, M (Br) = 80 g mol-1
E
0
AgBr / Ag = 0,073 V ; E
0
V
3+
/ V
2+
= - 0,256 V
2. Considere a seguinte célula, com resistência de 3,50 Ω e desprezando todas as
sobretensões
Pt⏐Fe2+
(0,10 M), Fe3+
(0,10 M), HClO4 (1 M)⏐⏐
Ce3+
(0,050 M), Ce4+
(0,10 M), HClO4 (1 M)⏐Pt
2.1 Calcule o potencial da célula se ela produzir uma corrente de 30,0 mA.
2.2 Calcule o potencial que tem que ser aplicado para que esta célula funcione
como electrolítica.
E
0
Ce
4+
/ Ce
3+
= 1,70 V ; E
0
Fe
3+
/ Fe
2+
= 0,767 V
3. Considere que a célula galvânica do problema anterior produz uma corrente de
100 mA quando nela existem as seguintes condições experimentais:
[Fe2+
]s=0,050 M, [Fe3+
]s=0,160 M, [Ce3+
]s=0,180 M, [Ce4+
]s=0,070 M. Considerando a
queda óhmica e a sobretensão de concentração, determine o potencial da célula.
R= 0,533 V](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-79-320.jpg)


















![Métodos voltamétricos
Análise Instrumental II 93
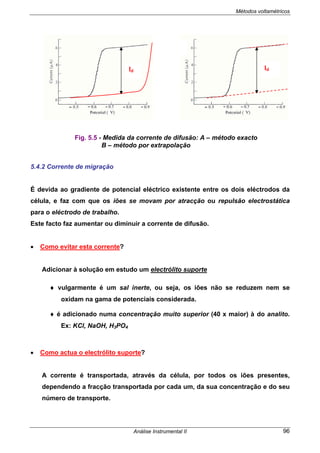
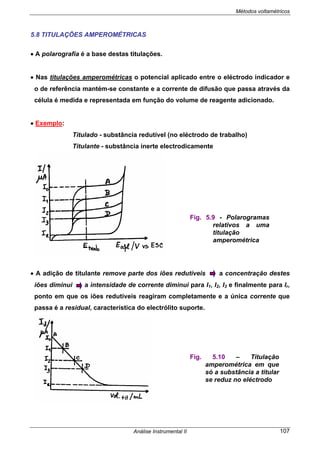
5.3 POLAROGRAMA
As curvas corrente/potencial, designadas por polarograma ou onda polarográfica,
são traçadas nas seguintes condições:
• Solução não agitada
• Apenas corrente de difusão
• Consideremos que o polarograma da fig. 5.4 b) diz respeito ao doseamento de
uma solução de Cd2+
. Neste caso o eléctrodo de trabalho funciona como cátodo.
Para Eapl < Ed verifica-se uma pequena corrente residual.
Para Eapl > Ed:
- dá-se a redução dos iões Cd2+
Cd2+
+ 2e-
+ Hg ↔Cd(Hg)
- rapidamente a [Cd2+
] à superfície do eléctrodo de Hg diminui e torna-se
nula (C0=0)
Fig. 5.4 - Polarogramas obtidos com a) DME (EGM) clássico b) MME.
a)
Ed E1/2 E /V vs ESC
b)](https://image.slidesharecdn.com/analise2ap-140313083959-phpapp02/85/analise-instrumental-98-320.jpg)













1) O documento introduz os métodos electroanalíticos, discutindo células electroquímicas, potenciais de célula, e tipos de correntes iônicas. 2) A seção sobre condutimetria explica condutância, condutividade específica e equivalente, e como a condutividade varia com a concentração e interações iônicas. Métodos de análise condutimétrica incluem titulações. 3) A potenciometria é abordada, incluindo células potenciomé





























![Potenciometria[1]](https://cdn.slidesharecdn.com/ss_thumbnails/potenciometria1-110613175045-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)





































