Baixado 648 vezes












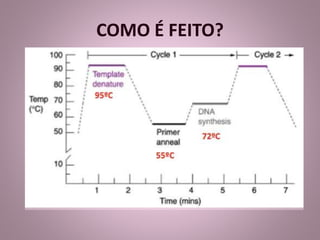




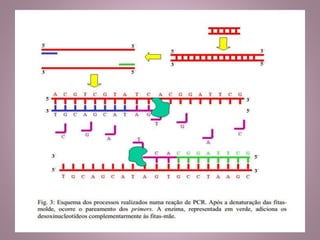









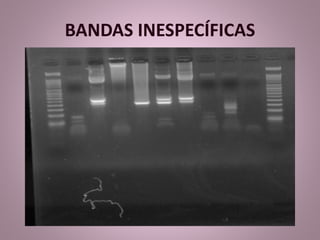







O documento descreve a técnica de Reação em Cadeia da Polimerase (PCR), seu histórico, componentes e etapas do processo. A PCR permite a amplificação de sequências específicas de DNA, utilizando primers e uma enzima polimerase, e envolve etapas como desnaturação, anelamento e extensão. Apesar de suas vantagens, a técnica tem limitações, como a necessidade de conhecer a sequência-alvo e a possibilidade de contaminação.




















































































