Baixado 57 vezes










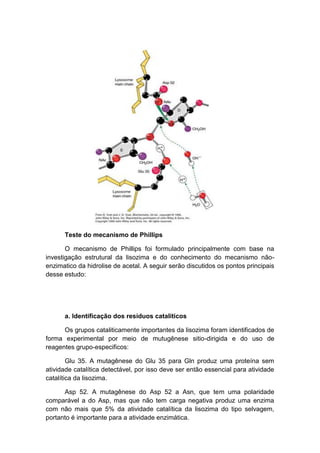



1) O documento discute os mecanismos de catálise enzimática, incluindo catálise ácido-básica, covalente, por íons metálicos e eletrostática. 2) A catálise enzimática é altamente eficiente porque as enzimas orientam os substratos de forma a estabilizar o estado de transição da reação e aumentar a velocidade da reação. 3) A lisozima é uma enzima descoberta por Alexander Fleming em 1922 que quebra a parede celular de bactérias.






















































































