Baixado 39 vezes


![Fármaco 2
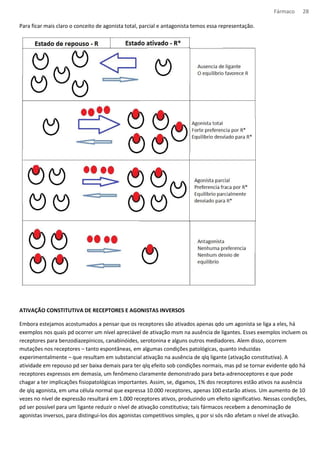
*Teste de bioequivalência consiste na demonstração de que o medicamento genérico e seu respectivo medicamento
de referência (aquele com o qual foi efetuada pesquisa clínica para comprovação da eficácia e segurança antes do
registro) apresentam a mesma biodisponibilidade¹ no organismo. A bioequivalência, na grande maioria dos casos,
assegura que o medicamento genérico ou similar apresente a mesma eficácia clínica e a mesma segurança em
relação ao produto de marca.
¹ Biodisponibilidade - é uma medida da extensão de uma droga terapeuticamente ativa que atinge a circulação
sistêmica e está disponível no local de ação. Indica a velocidade e a extensão de absorção de um princípio ativo em
uma forma de dosagem, a partir de sua curva concentração/tempo na circulação sistêmica ou sua excreção na urina.
(Biodisponibilidade é a concentração do medicamento disponível no sangue).
•Biodisponibilidade Adequada: Refere-se à fração de uma dose ingerida de uma droga que tem acesso à circulação
sistêmica, ou seja, corresponde a quantidade de fármaco disponível no organismo capaz para exercer efeito
terapêutico.
•Reação adversa: É qualquer resposta a um medicamento que seja prejudicial, não intencional, e que ocorra nas
doses normalmente utilizadas em seres humanos para profilaxia, diagnóstico e tratamento de doenças, ou para a
modificação de uma função fisiológica.
•Efeito colateral – Efeito associado ao medicamento, que pode ser o objetivo principal do remédio. Ex.: AAS é um
analgésico, antitérmico (antipirético), AINE, utilizado para dor. Um dos efeitos associados é a diminuição da
agregação plaquetária.
O alcance do efeito terapêutico depende das vias de administração, das características fisiológicas do
paciente e do próprio PA do medicamento. Para observar o efeito terapêutico, é ideal que as [plasmáticas]
ultrapassem a faixa de [] a partir da qual o efeito terapêutico será observado. Acima dessa faixa toxicidade. Abaixo
dessa faixa efeito subterapêutico.
[plasmática]
Obs.: Reação adversa e efeito colateral ocorrem na janela terapêutica.
Tempo
Toxicidade
Janela
terapêutica
[Subterapêutica]
Biodisponibilidade
adequada
Eliminação
Para um medicamento
ser seguro a janela
terapêutica deve ser
larga!](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-2-320.jpg)
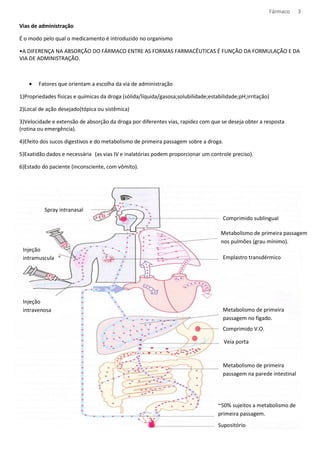
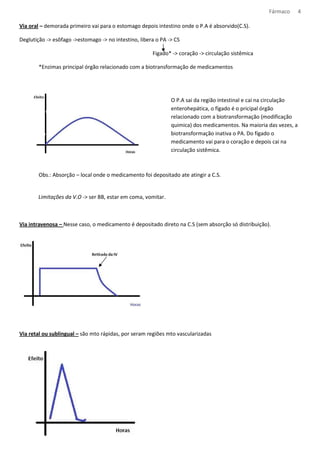

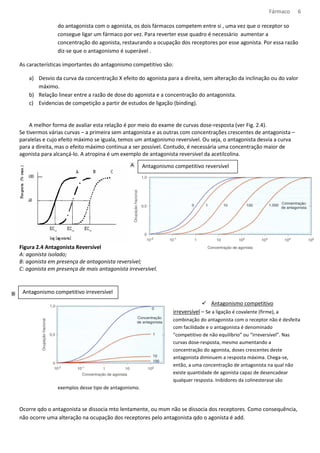
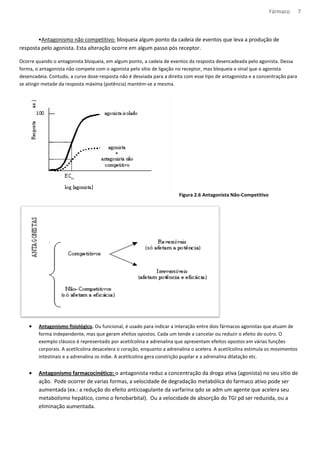


![Fármaco 10
Os medicamentos tem natureza acida ou básica, e podem ser encontrados na forma ionizada ou molecular,
dependendo do pH do meio.
Formula molecular – mais absorvido
Formula Ionizada – tem dificuldade de atravessar a bicamada lipídica
Meio com: [H+] = pH pH= característica do meio
[H+] = pH pka= caract. do composto
Equação de Handerson- Hasselbach
meio substancia
Exercicios
ion
molecular
1)Levando em consideração apenas os valores de pH do meio analise a seguinte situação:
Foi adm V.O um comprimido de AAS cuja pka é 3.0, determine qual o local onde esse PA será melhor ab
estomago (pH= 2,0) ou no intestino (pH= 6.0)?
Resp.:
= -1 - 10-1 =
No intestino (pH=6.0)
2 = 3 + - 2 – 3 =
6
no estomago
- = 1/10.
– 3 = - = 3
absorvido, no
- -1 =
- = 103 ou
Resp.: levando em consideração apenas o pH do meio, o AAS será absorvido de modo mais intenso pelo estomago.
____________________________________________________________
______________________________________________________________________________________________
__________________________________
1000
1](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-10-320.jpg)





![Fármaco 17
endógenos com atividade semelhante à da morfina. Hoje, sabe-se de uma família de peptídeos que são denominados encefalinas ou
endorfinas. A morfina, simplesmente, simula a ação dessas substâncias.
FISIOLOGIA DA PLACA TERMINAL
Receptores são macromoléculas (proteínas, em sua maioria) presentes nos tecidos e que se combinam quimicamente com os
fármacos de maneira relativamente específica. Isto é, fármacos interagem apenas com alguns receptores e vice-versa. A placa
terminal de uma fibra muscular esquelética, por exemplo, contém uma grande quantidade de receptores com afinidade para o
neurotransmissor acetilcolina.
Cada um desses receptores, que são chamados nicotínicos, é parte de um canal na membrana pós-sináptica que controla o
movimento intracelular de íons Na+. Em repouso, esta membrana pós-sináptica é relativamente impermeável ao Na+. Contudo,
quando o nervo é estimulado, ele libera, na placa terminal, acetilcolina que combina-se com os receptores nicotínicos e modifica-os
de tal forma que os canais se abrem e o Na+ flui para o interior da célula muscular. Quanto mais acetilcolina existir na região da
placa terminal, mais receptores serão ativados e mais canais se abrirão. Quando o número de canais abertos atinge um nível crítico
e o Na+ entra com rapidez suficiente para perturbar o equilíbrio iônico da membrana, ocorre uma despolarização localizada. Essa
despolarização localizada dispara a ativação de grande número de canais de Na+dependentes de voltagem e gera a despolarização
conduzida, conhecida como potencial de ação. O potencial de ação provoca a liberação – para o citosol – de Ca+2 a partir de seus
locais de ligação intracelular (particularmente, retículos endoplasmáticos e mitocôndrias). Este Ca+2 interage com proteínas
contráteis, gerando um encurtamento da célula muscular.
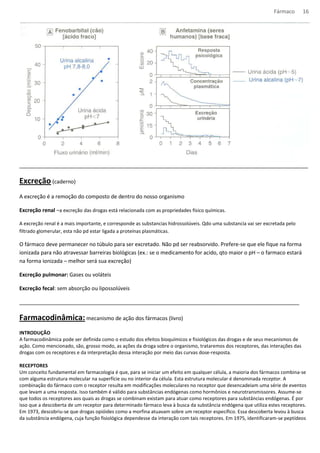
Figura 2.1 Receptor Nicotínico
O receptor nicotínico, por exemplo, é composto de 5 subunidades (duas α, uma β, uma γ e uma δ) que circundam uma depressão
central, que corresponde ao canal transmembranoso de Na+. Quando a acetilcolina se liga ao receptor (na subunidades α), o canal
central é aberto, permitindo a passagem de Na+.
Outros receptores – que não são canais iônicos – desencadeiam uma cascata de eventos graças à ação de segundos mensageiros. Os
fatores chave em muitos desses sistemas de segundos mensageiros são as proteínas G (há vários tipos). Essas proteínas hidrolizam o
trifosfato de guanosina (GTP) a difosfato de guanosina (GDP). As proteínas G transmitem a ativação de vários receptores a uma
etapa seguinte em uma série de reações. Em muitos casos, a etapa seguinte envolve a enzima adenilciclase. Vários hormônios,
fármacos etc. estimulam ou inibem a adenilciclase em vários tipos de receptores através das proteínas G diversas (inibitória ou
estimulatória). A adenilciclase catalisa a transformação de ATP em AMPc.
O AMPc ativa enzimas chamadas quinases que irão fosforilar diversas proteínas, resultando na resposta celular como abertura de
canais Ca+2 e ativação de outras enzimas. As proteínas G podem, também, ativar outras enzimas ou agir diretamente em canais
iônicos. Os receptores para adrenalina e noradrenalina são acoplados à proteína G.
_______________________________________________________________________________________________
VARIÁVEIS DA FARMACODINÂMICA
Afinidade. Mede a força de ligação entre droga e receptor e é determinada pelos tipos e número de ligações químicas. Reflete a
tendência de um fármaco se ligar ao receptor.
Eficácia. Ou “efeito máximo”, é a resposta máxima produzida pelo fármaco. Depende de quantos complexos fármaco-receptor são
formados e da eficiência com que o receptor ativado produz a ação celular. Ou seja, enquanto a afinidade é a tendência de um
fármaco se ligar ao receptor, a eficácia é a tendência de, uma vez ligado, esse fármaco modificar a função do receptor
desencadeando uma resposta. Independentemente da concentração do fármaco, atinge-se um ponto além do qual não ocorre mais
nenhum incremento na resposta. Tem-se, aí, resposta ou efeito máximo.
Potência. Ou sensibilidade, é a medida de quanto fármaco é necessário para desencadear uma determinada resposta. Quanto
menor a dose necessária para gerar tal resposta, mais potente é o fármaco. É calculada pela dose de fármaco que desencadeia 50%
da resposta máxima (EC50 [effective concentration 50%] ou DE50). Em geral, os fármacos de alta potência apresentam alta afinidade
pelos receptores, ocupando uma proporção significativa destes, mesmo em baixas concentrações.](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-17-320.jpg)


![Fármaco 25
Normalmente, o numero de molecs de adrenalina aplicadas ao tecido excede o numero de receptores (N), de modo q a
reação de ligação (NX) não reduz a concentração de adrenalina. A resposta produzida pela adrenalina será relacionada
com o numero de receptores ocupados.
A (droga) + R (receptor) = AR (complexo)
AFINIDADE E EFICÁCIA
· Afinidade - Capacidade de se associar, Avaliada pela EC50
· Eficácia - Capacidade de disparar uma resposta, Avaliada pelo Efeito máximo.
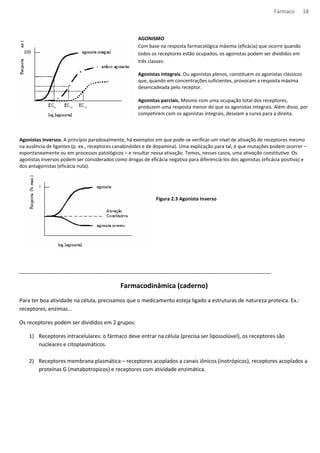
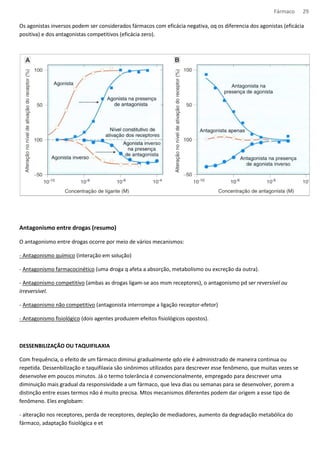
RELAÇÃO TEÓRICA ENTRE A OCUPAÇÃO E A CONCENTRAÇÃO DE LIGANTE
A constante de equilíbrio KA, é uma característica da droga e do
receptor; possui numericamente concentrações iguais da droga
para ocupar 50% dos sítios em equilíbrio.
Quanto maior a afinidade da droga pelos receptores, menor o
valor de KA.
A equação descreve a relação entre ocupação e concentração da
droga e produz uma curva característica conhecida como
hipérbole retangular (A), no trabalho farmacológico é comum
utilizarmos uma escala logarítmica de concentração, que
converte a hipérbole numa curva sigmoide (B).
CURVA DE CONCENTRAÇÃO DO AGONISTA DE EFEITO
A ligação das drogas a seus receptores nos tecidos pd ser medida diretamente. Entretanto, trata-se de uma resposta
biológica normal (como elevação da PA, relaxamento ou contração de um músculo e etc).
Essa ligação (droga-receptor) é medida e plotada como curva de concentração e efeito ou dose resposta.
Essa curva não pode ser utilizada para medir afinidade de drogas agonitas pelos seus receptores, visto que a resposta
produzida não é proporcional à ocupação dos receptores.
Todos os efeitos finais observados por drogas são um conjunto de alterações fisiológicas, a outra dificuldade em se
analisar a afinidade de uma droga pelo seu receptor é que a [droga] nos receptores é desconhecida, uma vez que os
agonistas podem ser sujeitos a degradação enzimática e etc.](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-25-320.jpg)
![Fármaco 31
2ª parte
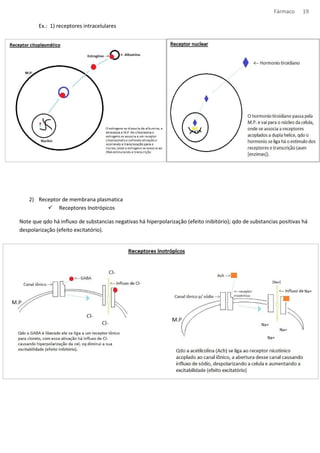
Alguns conceitos sobre Ca2+
Contração muscular
· A contração muscular ocorre em resposta a um aumento do [ca2+ ]i
· No músculo esquelético, a despolarização causa rápida liberação de ca2+ do reticulo sarcoplasmático (RS); no
músculo cardíaco, o ca2+ entra através de canais controlados por voltagem, e essa entrada inicial desencadeia a
liberação adicional do RS; no músculo liso, o sinal de ca2+ se deve parcialmente a entrada de ca2+ e parcialmente
a liberação do RS mediada pelo IP3.
· No músculo liso, a contração pd ocorrer sem potenciais de ação, por exemplo qdo um agonista nos receptores
acoplados a proteína G leva a formação de IP3.
· A ativação do mecanismo contrátil do músculo liso envolve a fosforização da cadeia leve da miosina, um
mecanismo regulado por uma variedade de sistemas de segundos mensageiros.
Coração
Hiperpolarização
Despolarização
Efeito inibitório
Efeito excitatório
Influxo de anions (-) Cl-
Efluxo de cátions (+)k+, k+ , k+
Influxo de cátions (+)
Na+,Na+, Na+
Neurônio
Musc. liso](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-31-320.jpg)
![Fármaco 32
Liberação de mediadores
· A maioria dos mediadores químicos é armazenada em vesículas e liberada por exocitose. Alguns são
sintetizados em função da demanda e são liberados por difusão ou através de carregadores presentes
na membrana plasmática.
· A exocitose ocorre em respostas a um aumento no [ca2+]i resultante da interação mediada pelo ca2+
entre proteínas da vesícula sináptica e da membrana plasmática, fazendo com que as membranas se
fundam.
· Após liberar seu conteúdo, as vesículas são recicladas e novamente preenchidas com transmissor.
· Muitas cels secretoras contem mais de um tipo de vesícula, preenchidas com diferentes mediadores e
secretadas independentemente.
· Os mediadores armazenados (por ex.: neurotransmissores) podem se liberados diretamente no citosol,
independentemente do ca2+ e da exocitose, por fármacos que interagem com os mecanismos de
transporte da membrana.
· Os mediadores que não são armazenados, como os prostanóides e o oxido nítrico, são liberados através
do aumento de [ca2+]i, que ativa as enzimas responsáveis pela sua síntese.](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-32-320.jpg)




![Fármaco 38
Uma importante característica das aminas simpatomiméticas de ação indireta consiste no desenvolvimento de
acentuada tolerância. Doses repetidas de anfetamina ou tiramina, por exemplo, produzem respostas pressoras
progressivamente menores. Isso provavelmente é causado por uma depleção das reservas liberáveis de noradrenalina.
Verifica-se também desenvolvimento de tolerância similar aos efeitos centrais com a administração repetida
explicando, em parte, a tendência das anfetaminas e fármacos relacionados a causar dependência.
Ações. As ações periféricas das aminas simpatomiméticas de ação indireta incluem broncodilatação, pressão arterial
aumentada, vasoconstrição periférica, aumento da freqüência cardíaca e da força de contração do miocárdio, e inibição
da motilidade intestinal. À exceção da efedrina, que ainda é algumas vezes utilizada como descongestionante nasal, em
virtude de sua ação central bem menos, esses fármacos não são mais usados por causa dos seus efeitos
simpatomiméticos periféricos.
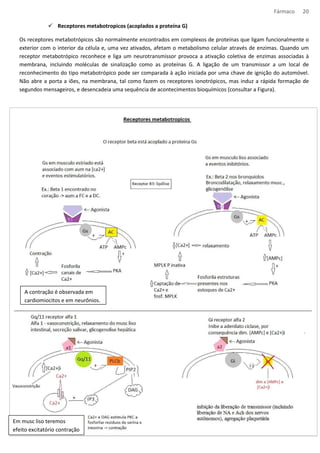
Farmacodinâmica das aminas de ação indireta
· Guanetidina – não tem uso na clinica. Inibe a liberação NE, ela entra pela captação 1, se acumula nas vesículas
liberando NE para o citoplasma e a MAO degrada-o. A diminuição de NE na fenda sináptica, causa
comprometimento do impulso nervoso.
· Anfetamina – entra pela captação 1 (pré-sinaptica), entra na vesícula de NE pelo carreador
monoaminovesicular. Entra anfetamina sai NE da vesícula e sofre endocitose, já que parte da MAO esta
“ocupada” degradando anfetamina. Com isso há o aumento da [NE] na fenda.
· Efedrina e tiramina – passa pelo mesmo processo da anfetamina (substrato para a MAO). A tiramina é
encontrada nos alimentos como qjo, embutidos, cervejas e etc. Nos enterocitos e hepatocitos há uma
quantidade alta de MAO por isso não temos uma crise hipertensiva.
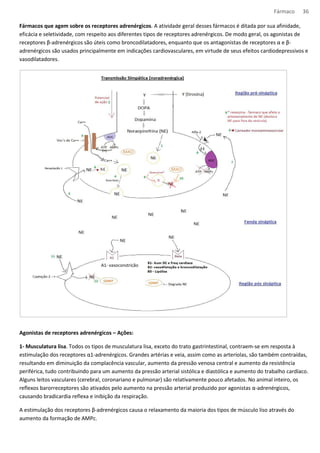
Locais de ação dos fármacos (MeNa – metilnoradrenalina; NA - noradrenalina).](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-38-320.jpg)



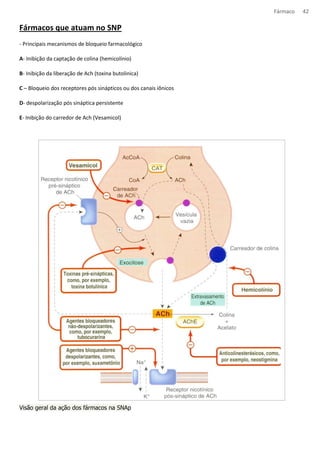









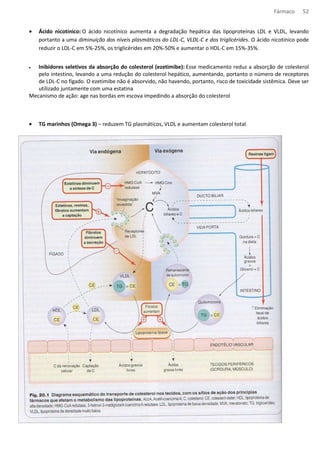
![Fármaco 53
Angina (cardiopatia isquêmica)
Ocorre angina qdo a oferta de O2 ao miocárdio é insuficiente para suas necessidades. A dor tem distribuição
característica no peito, membro superior e pescoço e é ocasionada por esforço físico, frio ou agitação.
A angina pd ser de três tipos:
- Angina estável – causada pelo esforço físico, é produzida por um aumento da demanda sobre o coração causada por
um estreitamento fixo dos vasos coronarianos, quase sempre por ateroma. A terapia sintomática é direcionada para
alterar o trabalho cardíaco usando nitratos orgânicos, antagonistas dos receptores beta adrenérgicos (não seletivos
entre b1 e b2) e/ou antagonistas de cálcio. Juntamente com o tratamento da doença ateromatosa subjacente,
geralmente incluindo uma estatina e profilaxia contra trombose com um antiplaquetário, geralmente aspirina.
-Angina instável – esta se caracteriza por dor que ocorre com, cada vez menos, esforço físico, culminando em dor de
repouso. A patologia é semelhante ao infarto do miocárdio, se tem aterosclerose sem oclusão completa do vaso o
intuito da terapia é reduzir o risco de infarto. A aspirina faz cair pela metade o risco de infarto, a heparina e os
antagonistas dos receptores de glicoproteinas plaquetarios fazem um acréscimo a esse beneficio.
- Angina variantes – esta é incomum, ocorre em repouso e é causada por espasmo coronariano, novamente, em geral,
associado a doença ateromatosa. A terapia é com vasodilatadores coronarianos (ex.: nitratos orgânicos, antagonistas de
cálcio).
Fármacos antianginosos
· Nitratos orgânicos – relaxam is músculos lisos vasculares e alguns outros (ex.: esofágico e biliares). Causam
acentuado relaxamento venoso, com uma consequente redução da pressão venosa central (redução da pré
carga).
Em resumo, a ação antianginosa dos nitratos envolve:
• Redução do consumo cardíaco de oxigênio, secundariamente redução da pré-carga e da pós-carga cardíaca.
• Redistribuição do fluxo coronariano em direção a áreas isquêmicas através de colaterais.
• Alivio do espasmo coronariano
Mecanismo de ação : os nitratos organicos são metabolizados com a liberação de oxido nítrico. Em concentrações
atingidas durante o uso terapêutico, isto envolve uma etapa enzimática e, possivelmente, uma reação com grupos
sulfidrila dos tecidos. O oxido nítrico ativa a guanilato ciclase solúvel, aumentando a formação de GMPc, que ativa a
proteína quinase G e leva a uma cascata de efeito no músculo liso, culminando em desfosforilação das cadeias leve da
miosina e sequestro de calcio intracelular, com consequente relaxamento.
Caderno
Mecanismo de ação - doadores de NO, penetra na cel do musc liso vasc, aum GMPc estimulando PTN PKC, diminuindo
[Ca++]i, causando vasodilatação (melhora a irrigação) dim PA e DC, dim a demanda de Oxig pelo coração.
Reação adversa – cefaleia vascular](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-53-320.jpg)
![Fármaco 54
· Ativadores dos canais de potássio (k+)- O nicorandil combina a ativação do canal de K com ações
nitrovasodilatadoras. É um dilatador arterial e venoso, causa os efeitos indesejáveis esperados como cefaleia,
rubor e tonturas. É usado em pacientes que continuam sintomáticos apesar de uma ótima conduta com outros
fármacos, mtas vezes enquanto aguardam cirurgia ou angioplastia.
Caderno
agem nos vasos, nas cels do musc liso vasc. Causa efluxo de K+, essa sobrecarga positiva esta relacionada com a
hiperpolarização, inibindo Voc’s de Ca++. Dim [Ca++] - vasodilatação. Ex.: nicorandil
· Antagonistas dos receptores beta adrenergicos (não seletivos)– são importantes na profilaxia da angina e no
tratamento de pacientes com angina instável. Funcionam para estas indicações, reduz o consumo cardíaco de
oxigênio. Ademais , reduz o risco de morte após infarto do miocardio, através de sua ação antiarritmica.
Caderno
Presente no miocárdio, dim o consumo de oxigênio pelo coração, dim DC, inibe a secreção de renina nos rins,
dim o tônus. Ex.: Propanolol, metropolol
· Bloqueadores do canais de cálcio (ou antagonistas de cálcio) – bloqueiam a entrada de cálcio, impedindo a
abertura dos canais de cálcio controlados por voltagem. Impedem espasmos e a vasoconstrição. Ex.: verapamil,
diltiazem.
Efeitos sobre a musculatura lisa – dilatação arteriolar generalizada
Efeito cardíaco – proteção isquêmica, vasodilatação e a dim do trabalho cardíaco reduzem a demanda
metabólica cardíaca.
Reação adversa- constipação, hipotensão postural, cefaleia.
__________________________________________________________________________________________________
Cardiotônicos
Cardiotônicos: Aumentam a força de contração e o DC, indicados no tratamento de Insuficiência Cardíaca..
Tratamento de ICC e aritimias cardíacas
Fisiologia da Insuficiência Cardíaca
Insuficiência Cardíaca: falha do coração em bombear sangue suficiente para suprir as necessidades de O2 e nutrientes
do organismo. Causando dim do DC, e excesso de trabalho para o coração.](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-54-320.jpg)
![Fármaco 55
Insuficiência cardíaca aguda:
↓DC
Baroreceptores
Taquicardia ↑ RVP ↑ Sistema RAA
Insuficiência Cardíaca (resposta tardia):
DC
Compensação renal (retenção de água e sódio)
Edema
Sinais e Sintomas: Taquicardia, ↓ tolerância exercício, dispnéia, edema periférico e pulmonar, cardiomegalia.
Medicamentos Cardiotônicos:
Digitálicos (inibição de na+/K+/ Atpase) – inibe a saída de [Na+]i despolarizando a fibra abrindo Voc’s de Ca++, que
estimula a saída de calcio do reticulo.
Ex.: digoxina, oubaina, digitoxina (deve-se evitar o uso desses medicamentos com cafeína pois potencializa seu efeito).
Efeitos: aum [Ca++] dentro do miocito, aum FC = ionotropismo positivo
Dim FC = cronotropismo negativo. Levam tbm a uma inibição da atividade simpática adrenérgica.
Não digitálicos – aum [Ca++]i sobre dois mecanismo:
- agonistas B1 (dopamina, dobutamina) aum a síntese de AMPc e produção de Ca++
- inibidores da fosfodiesterase (PDE) reduzem a degradação de AMPc. Ex.: milrirona
Efeitos – aum [AMPc]= aum o inotropismo, vasodilatação.
Dilatação coração
Edema pulmonar
Desoxigenação
Edema periférico](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-55-320.jpg)
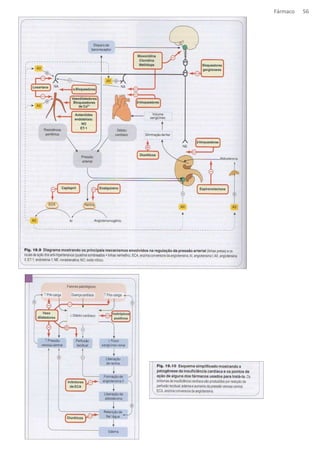






![Fármaco 64
Fármacos antiinflamatórios
Os mediadores químicos da inflamação são produzidos a partir de substancias de natureza lipídica e nem sempre estão envolvidas
com o processo de inflamação.
EX.: Prostagladinas (PG)
Tromboquixanos (TX)
Leucotrienos (LT)
Autacoides – não possuem lugar de síntese e degradação
Obs.: PG, TX e LT são produzidos pelo acido araquidônico (composto presente na membrana plasmática) pela ação das enzimas COX
e LIPOX.
Mecanismo de ação : membrana plasmática um dos componentes é Acido araquidônico (AA) que é liberado pela ação da fosfolipase
A2 (PLA2), libera AA no citoplasma deixando-o livre para ação de duas enzimas COX e a LIPOX.
COX - forma PG* e TX
LIPOX - forma LT ( hipersensibilidade e vasoconstrição).
* (dor, importante para as cels do estomago para proteção gástrica e para perfusão renal)
Isoformas da Cox
o COX 1 – reação fisiológicas (encontrada em plaquetas libera TXA2)
Seu bloqueio leva a dim de PG, prostaciclinas e TX
o COX 2 – processos inflamatórios, presentes em tecido lesado (encontrada endotelio vascular produção de PGI2 -
vasodilatação)
Seu bloqueio evita os efeitos indesejáveis da inflamação, mas aum [LT] promovendo broncoespasmo (indivíduos
asmáticos tem uma precipitação da crise asmatica).
o COX 3 – Encontrada em macrófagos
AINE
Eicosanoides
Eicosanóides são mediadores inflamatórios (que modulam a resposta inflamatória) de origem lipídica, sintetizados a
partir dos ácidos graxos ômega-6, como o ácido araquidônico (AA).Frente a um estímulo antigênico, AA, são
mobilizados da membrana de células imunes pela enzima fosfolipase A2. Esses ácidos graxos competem entre si pelas
mesmas vias enzimáticas (cicloxigenase e lipoxigenase) para a formação dos eicosanóides.
PGA2- formada a partir de PGH2, efeito vasodilatador e antiagregante.](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-64-320.jpg)

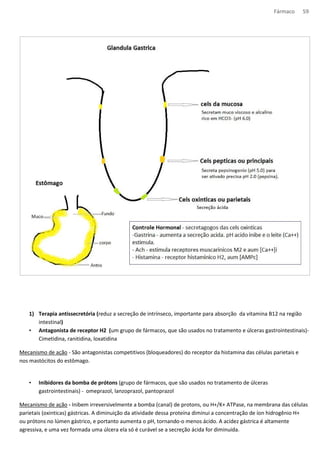
![Fármaco 66
MP
AA
PLA2
AA
Citoplasma
LIPOX COX
LT TG e TX
AINES
(-)
[LT] os AINES
(-)
AIES* *AIES – núcleo esteroidal.
Glicorticoides (cortisol) – aum a
glicemia
Mineralocorticoides – aum [Na+]
(aldosterona)](https://image.slidesharecdn.com/resumo-farmaco-completo-120625132441-phpapp02-140915100019-phpapp02/85/Resumo-farmaco-completo-120625132441-phpapp02-66-320.jpg)






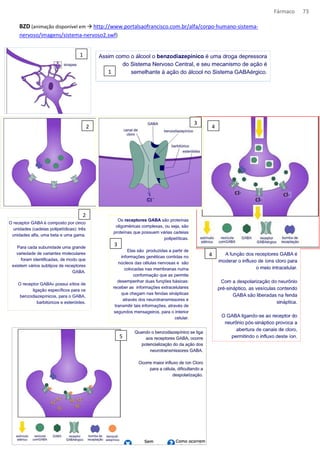

O documento discute conceitos fundamentais de farmacologia, incluindo: (1) a definição de termos como remédio, medicamento, fármaco e droga; (2) as vias de administração de medicamentos e seus efeitos na absorção e biodisponibilidade; (3) os tipos de interação entre fármacos, incluindo agonismo, antagonismo competitivo e não competitivo.









































































