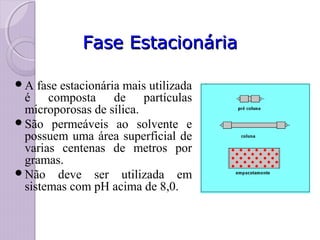
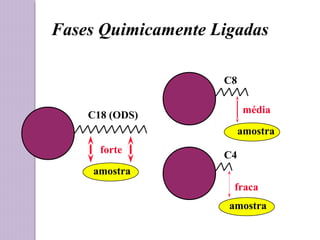
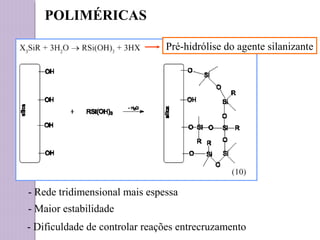
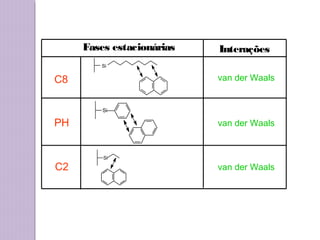
Baixado 537 vezes







































![Mudança no modificador orgânicoMudança no modificador orgânico
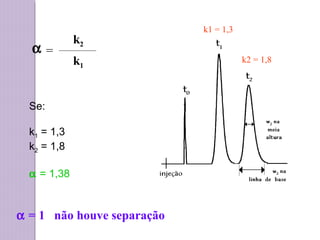
a
b
c
d
a
b
c
d
[ MeOH/H2O ]
par crítico : c,d
[ THF/H2O ]
par crítico : a,b
a
b
c
d
[ MeOH/THF/H2O ]](https://image.slidesharecdn.com/cromatografialiquida-130626084617-phpapp01/85/Cromatografia-liquida-63-320.jpg)








![ReferênciasReferências
TRINDADE, Magno Aparecido Gonçalves; RINALDO, Daniel;
VILEGAS, Wagner and ZANONI, Maria Valnice Boldrin.
Determinação de corantes marcadores do tipo azo e
antraquinona em combustíveis por cromatografia líquida
com detecção eletroquímica. Quím. Nova [online]. 2010,
vol.33, n.1, pp. 146-150. ISSN 0100-4042.
AFLATOXINAS EM ALIMENTOS DESTINADOS A
BOVINOS E EM AMOSTRAS DE LEITE DA REGIÃO DE
LAVRAS, MINAS GERAIS – BRASIL - Maria Marlucia
Gomes Pereira1, Eliana Pinheiro de Carvalho2, Guilherme
Prado3, Carlos Alberto da Rocha Rosa4, Thaís Veloso5,
Leandro Augusto Ferreira de Souza5,Jéssika Mara Martins
Ribeiro6](https://image.slidesharecdn.com/cromatografialiquida-130626084617-phpapp01/85/Cromatografia-liquida-76-320.jpg)

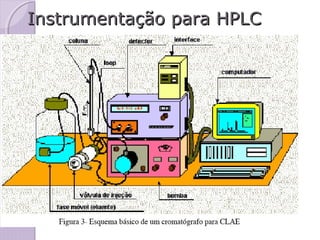
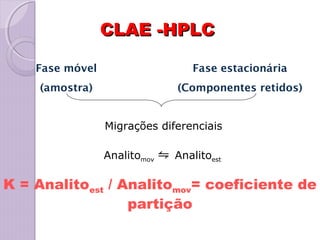
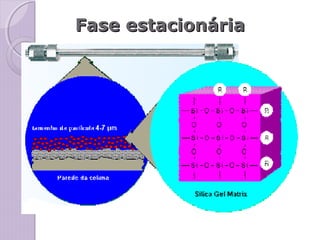
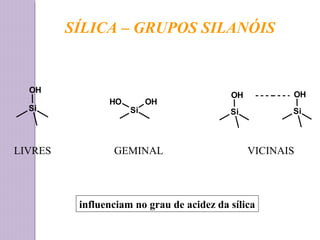
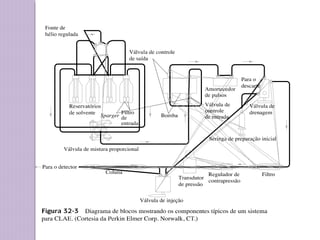
O documento descreve os princípios e componentes da cromatografia líquida de alta eficiência (HPLC). Explica que a HPLC envolve a separação de componentes de uma amostra líquida através da interação diferencial desses componentes com fases móvel e estacionária. Detalha os principais componentes de um cromatógrafo HPLC, incluindo a bomba, injetor, coluna e detectores, e discute os tipos comuns de fases estacionárias e modos de separação.


































![seminário HPLC[816].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/seminriohplc816-221218212347-3624f57f-thumbnail.jpg?width=640&height=640&fit=bounds)



![Analítica SLIDE (1) OFC[1].pptx aula de quimica analitica](https://cdn.slidesharecdn.com/ss_thumbnails/analticaslide1ofc1-250930111752-44d614a6-thumbnail.jpg?width=640&height=640&fit=bounds)
















































