

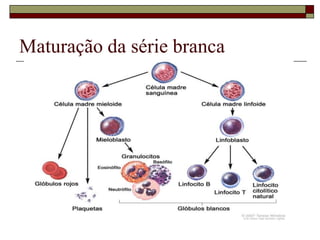
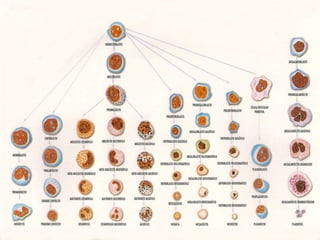


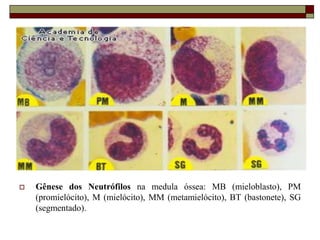

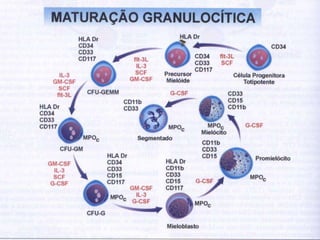







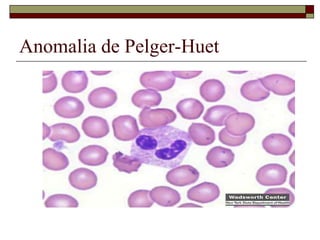
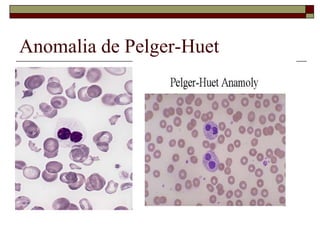

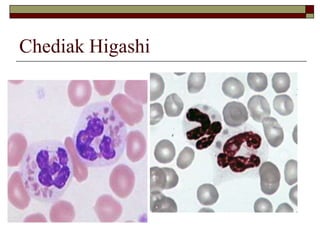
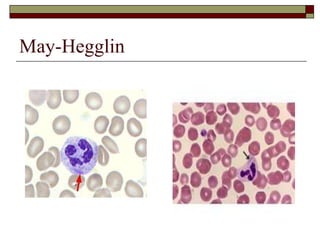




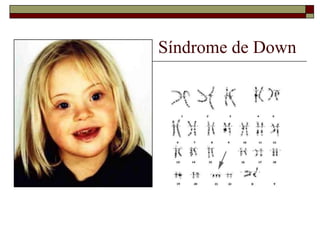

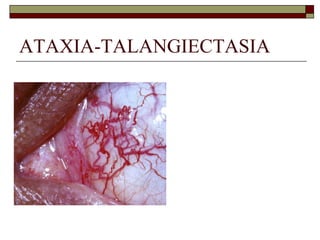





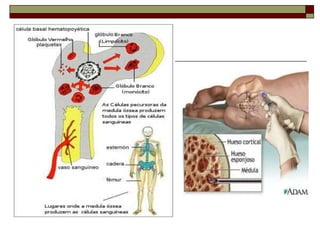















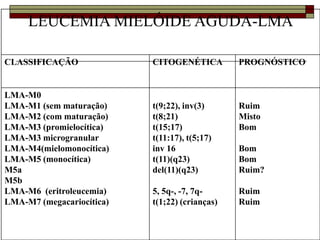
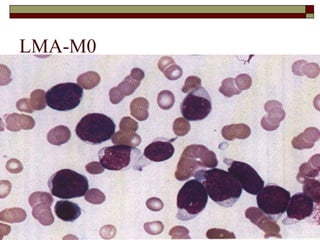













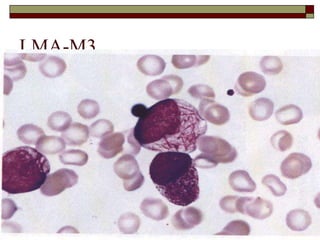
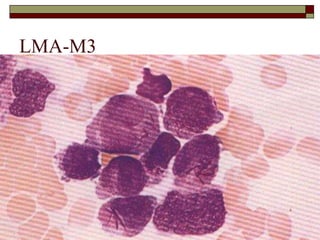

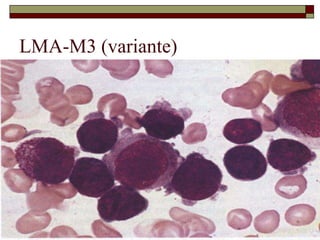

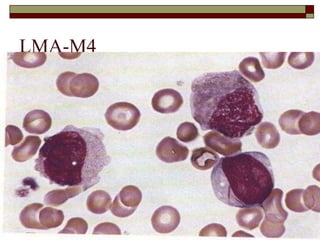
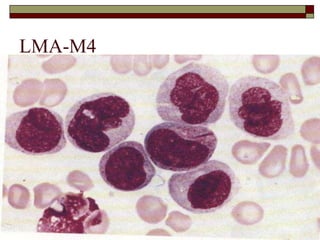














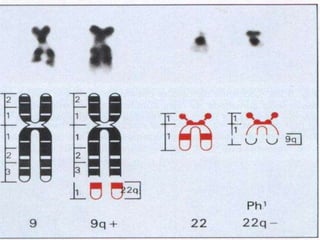


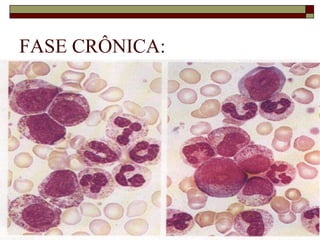
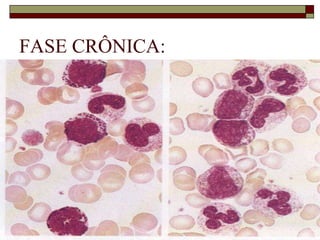













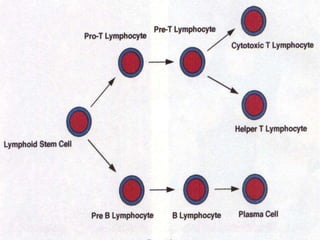








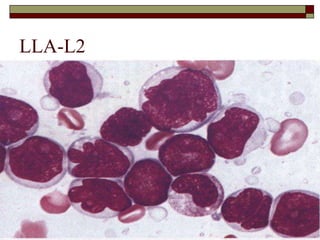





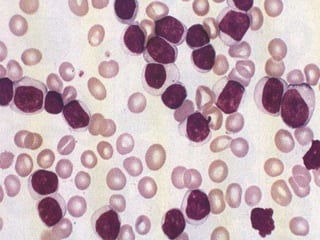
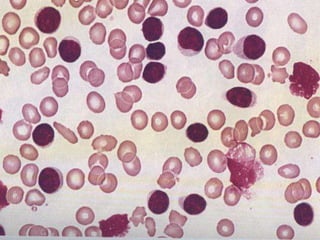
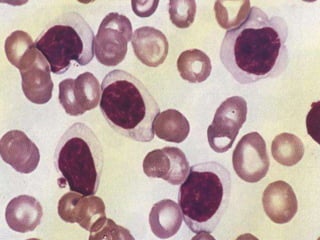

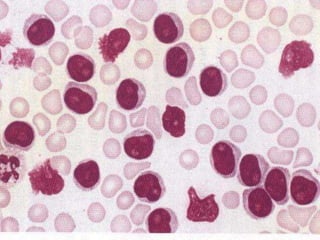
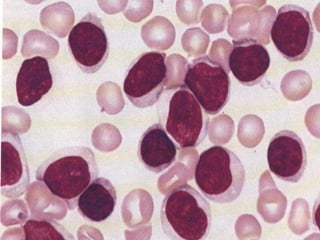
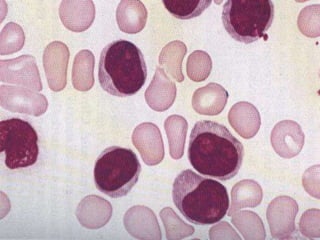

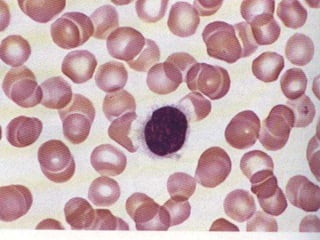
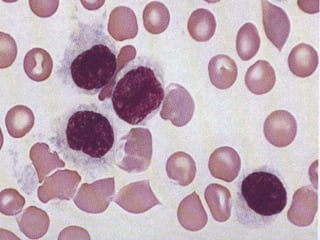



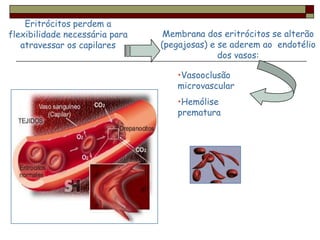







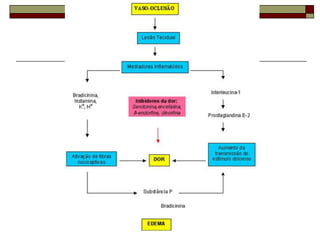


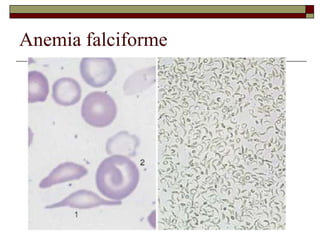
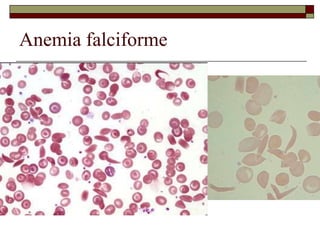
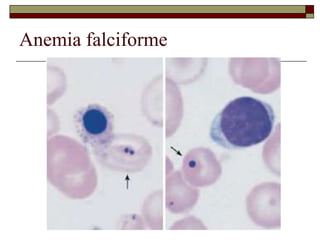








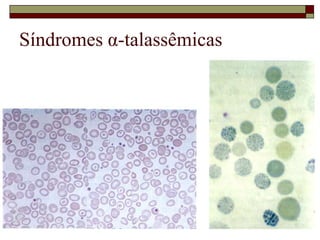





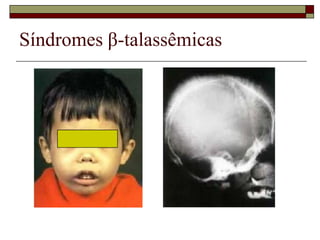
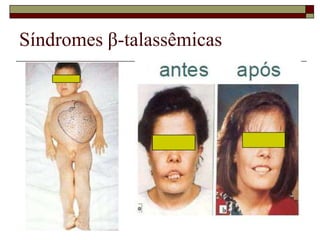

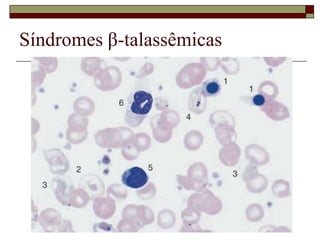
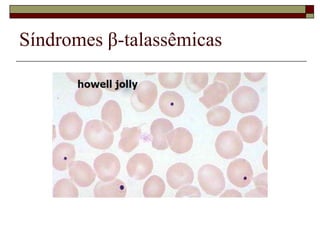

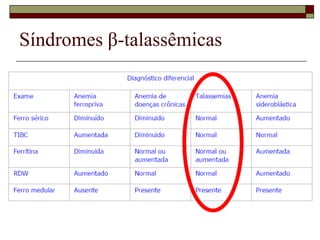




O documento descreve as principais características da maturação da série branca do sangue e das leucemias mielóides agudas. Resume os principais tipos de leucemia mielóide aguda de acordo com a Classificação FAB, incluindo sinais, sintomas, recursos diagnósticos e aspectos citogenéticos e prognósticos.






























































