Baixado 111 vezes








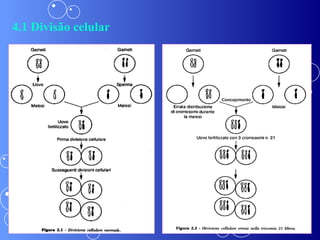
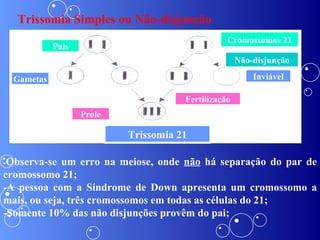
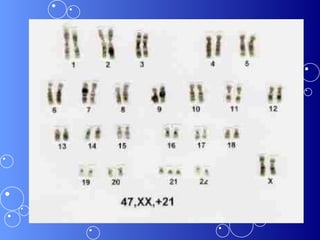


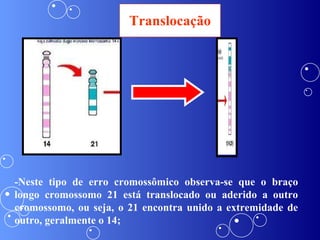


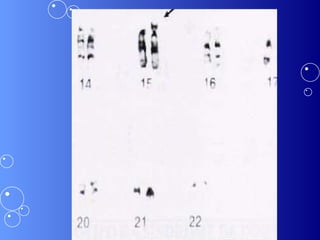




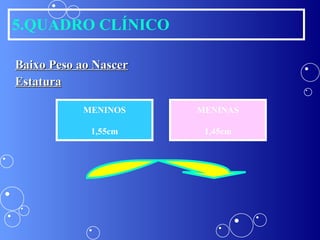
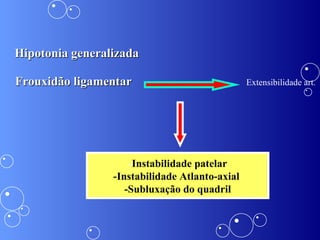











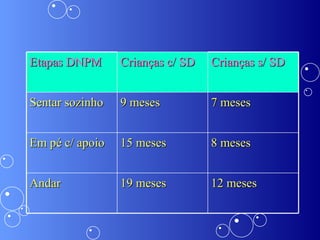






















A síndrome de Down é uma anomalia genética causada pela presença de um cromossomo 21 extra. Isso resulta em atraso no desenvolvimento motor e mental da criança. Características comuns incluem face achatada, olhos oblíquos, orelhas pequenas e problemas de saúde como cardiopatias e problemas auditivos. O diagnóstico é feito através de exames pré-natais ou cromossômicos após o nascimento. O tratamento envolve uma equipe multidisciplinar e fisioterapia foc
![[c7s] Síndrome de Down](https://cdn.slidesharecdn.com/ss_thumbnails/sindromededown-111129143208-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)














































