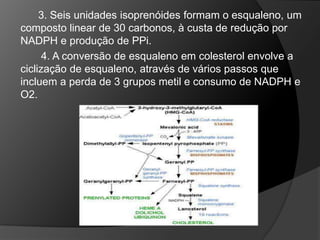
Os lipídios desempenham papéis cruciais no metabolismo energético e são transportados na corrente sanguínea por lipoproteínas. Dislipidemias, que afetam os níveis de lipídios no sangue, são fatores de risco para doenças cardiovasculares e podem ser primárias ou secundárias, relacionadas a hábitos de vida ou condições médicas. Erros no metabolismo do colesterol e ácidos biliares podem resultar em várias doenças genéticas, com manifestações clínicas significativas.