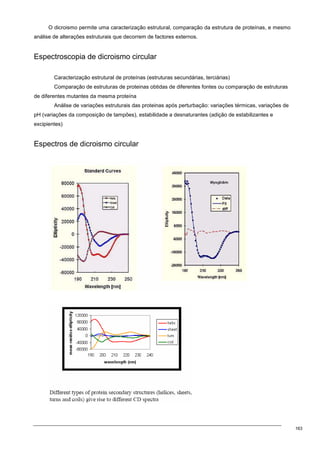
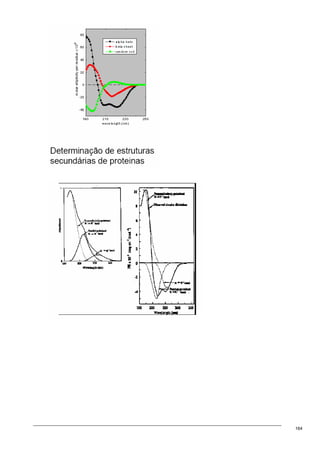
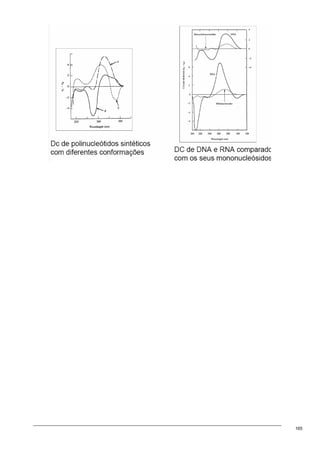
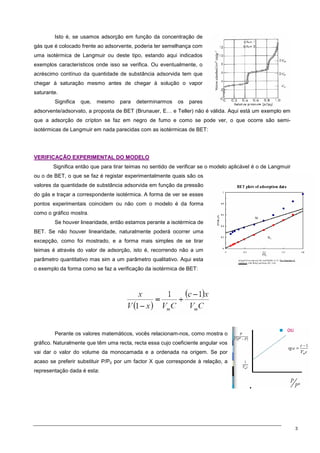
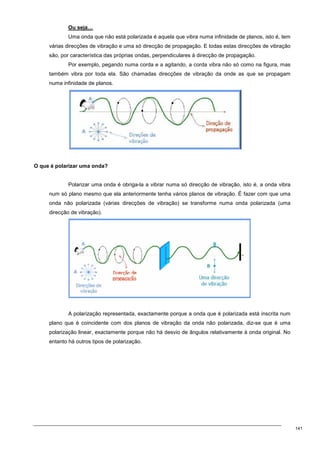
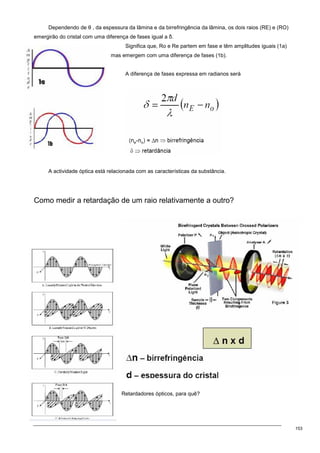
O documento discute os modelos de Langmuir e BET para adsorção em interfaces sólido-gás. Explica que o modelo de Langmuir se aplica quando há formação de uma única camada molecular, enquanto o modelo BET é mais geral e se aplica quando há formação de múltiplas camadas. Também descreve métodos experimentais para verificar qual modelo se aplica e medir a área superficial de sólidos.
























![25
Como permutador fracamente ácido temos o RCO2
-
, esse já só permitem separação quando os
pH são superiores aos valores de pK da substância a determinar.
No caso de se tratarem de permutadores aniónicos também eles poderão ser fortemente básicos
e também têm a vantagem de poderem ser utilizados num reino completo de pH, ou então fracamente
básico que permutam só a valores de pH inferiores ao valor de pKa.
E
EQ
QU
UI
IL
LÍ
ÍB
BR
RI
IO
O D
DE
E D
DO
ON
NN
NA
AN
N
O equilíbrio que se dá numa resina, se considerarmos um permutador catiónico na forma do ião
Na
+
(catião sódio), e se quisermos passar uma solução envolvendo iões Li
+
, o que vai acontecer é que o
sódio vai permutar temporariamente com o lítio e estabelece-se um equilíbrio cuja constante de equilíbrio
no fundo determina o coeficiente de selectividade da própria resina, isto é, diz-nos qual a capacidade que a
resina tem de fixar o lítio em substituição do sódio, em maior ou menor extensão do que a fixação do sódio
à própria resina. Este equilíbrio dá-se quando as concentrações dos iões no eluente são superiores à
concentração do ião na própria resina e determina o chamado equilíbrio de Donnan, que representa o
equilíbrio de cargas que existe quer à superfície quer no exterior da resina.
É este equilíbrio que se passa ao nível das nossas células. As trocas de sódio/potássio nas
nossas células (que determinam a bomba sódio/potássio que tem implicações no funcionamento cardíaco
[ritmo cardíaco, etc.]) estão dependentes de uma permuta iónica que é definida através de um equilíbrio
que acaba por determinar a concentração de iões fora das células e no interior. Há formas de alterar este
equilíbrio, quer por medicação, quer por outros processos cíclicos.
Exactamente porque a carga interna da resina é considerável, a possibilidade de atraque de iões
à resina é favorecida. Se tiver um ambiente iónico sedento de cargas, é mais fácil de captar iões do que de
rejeitá-los, e por isso ocorre o tal fenómeno de troca iónica: o equilíbrio de Donnan.
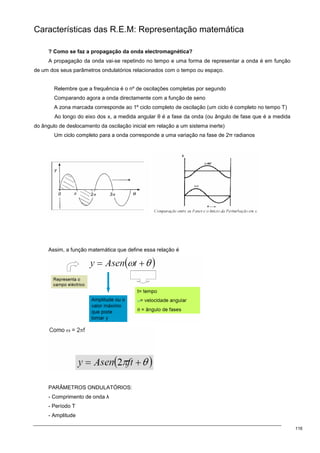
O equilíbrio de Donnan pode ser representado como aqui está indicado:
Se considerarmos um
permutador aniónico fortemente básico
que está na fórmula de anião Cl
-
(isto é,
temos um polímero de resina e na ponta
temos ligados iões cloreto) e se
quisermos passar uma solução de cargas
Cl
-
, o balanço de cargas que ocorre dentro
e fora do permutador, a concentração de
R
+
da resina mais a concentração de iões
K
+
, tem de ser exactamente igual à
concentração de Cl
-
no interior da resina, isto porque tem de haver electroneutralidade e só assim se
entende que as cargas positivas no interior da resina se igualam às cargas negativas. No exterior da resina
também tem de haver electroneutralidade, está a passar KCl, então a concentração fora da resina de K
+
é
igual à de Cl
-
.](https://image.slidesharecdn.com/sebentafisicaaplicda-230208235124-4cc330c5/85/sebenta-FisicaApliCada-pdf-25-320.jpg)













![39
se ele for negativo → há adsorção é positiva, logo há presença de um agente
tensioactivo.
Ex.: Os sabões, os ácidos gordos, as aminas, as próprias proteínas têm funções
tensoras.
Se ele for positivo → há uma adsorção negativa.
Ex.:os açúcares, os polissacarídeos ou qualquer coisa que se dissolva numa solução.
V
VA
AR
RI
IA
AÇ
ÇÕ
ÕE
ES
S D
DA
A T
TE
EN
NS
SÃ
ÃO
O S
SU
UP
PE
ER
RF
FI
IC
CI
IA
AL
L E
EM
M F
FU
UN
NÇ
ÇÃ
ÃO
O D
DA
A C
CO
ON
NC
CE
EN
NT
TR
RA
AÇ
ÇÃ
ÃO
O D
DE
E T
TE
EN
NS
SI
IO
OA
AC
CT
TI
IV
VO
O
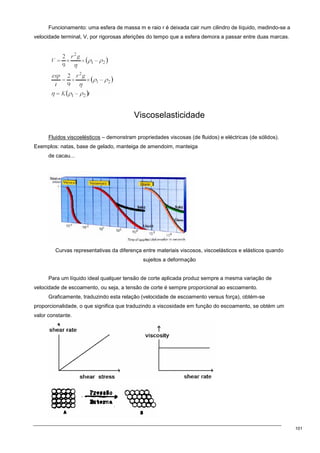
Três tipos de comportamento de substâncias.
Tipo 1 → aumento da tensão superficial em função da
concentração e são substâncias (compostos orgânicos) que normalmente
têm grupos OH ou COOH e um grupo apolar;
Tipo 2 → decréscimo da tensão é uma coisa muito atenuada; Ex.:
sais inorgânicos e sacarose
Tipo 3 → decréscimo brutal da tensão até atingir a concentração
micelar crítica e depois uma estabilização da mesma. Ex.: Sais orgânicos de cadeia média (sabões,
RCOONa+); sais de amóni quaternário [(CH3)3RN+Cl -]; compostos de polioxietileno [R(OCH2CH2)nOH
em que n=5-15]
↓ Os grupo funcionais, signicam:
que se se fizer um sitema constituido por três fases, em que por exemplo temos água, noutro
fenol e finalmente, noutro temos uma substância tensora, isto é uma mistura de água e fenol → Se se
tentar dissolver fenol em água, até uma determinada concentração, temos uma fase única, isto é, o fenol
solubiliza-se em água.:
Até cerca de 7% de fenol há uma fase homogénea porque todo o fenol se solubiliza em água.
A partir de 7% há a formação de duas fase, não é possível dissolver mais fenol em água. Só é
possível através da adição de um agente tensioactivo.
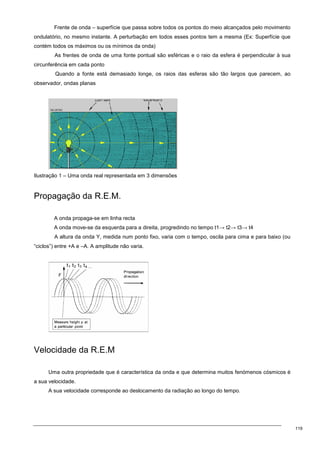
Atenção tentar perceber pelo gráfico….
E então se se considerar que aqui está
fenol e aqui está água, vai-se ter uma zona → esa
zona daqui é uma zona perfeitamente
homogénea, depois vou ter uma zona que aqui
está representada por isto, uma zona onde não há
solubilização, há a formação de duas fases. Mas
quando o fenol passa a estar em muito maior
quantidade que a água, ocorre outra vez a
formação de uma fase homogénea. Agora, se eu me localizar aqui, eu posso transformar esta fase
heterogénea em homogénea se eu adicionar progressivamente quantidades de agente tensioactivo.
Se eu for aumentando desde 0 a 100% a quantidade de agente tensioactivo, eu atingo uma fese
homogénea. Claro que o mesmo já não acontece aqui. Eu posso aumentar, passo por uma fase
homogénea mas logo a seguir passo por uma fase heterogénea.](https://image.slidesharecdn.com/sebentafisicaaplicda-230208235124-4cc330c5/85/sebenta-FisicaApliCada-pdf-39-320.jpg)























































![97
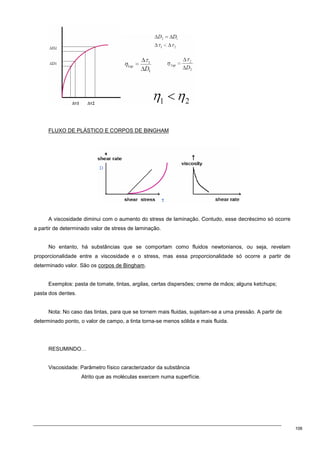
Viscosidade: é função de ….
1. Forma e estrutura do soluto
2. Peso molecular do soluto
3. Concentração do soluto
4. Temperatura
5. Pressão
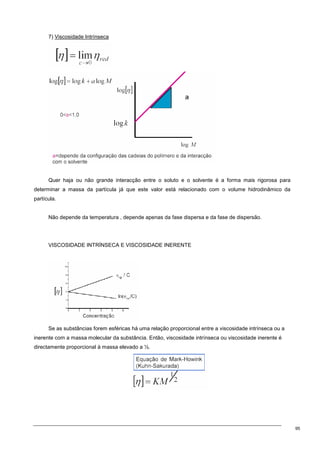
1.Viscosidade estrutura da partícula
Devido às inúmeras conformações que a cadeia polimérica pode assumir, a melhor representação
da sua morfologia é a de um novelo aleatório.
A dependência de [η] com o formato molecular deve-se ao movimento de rotação da molécula, ou
seja, ao coeficiente de fricção que cada segmento do polímero exerce sobre o centro de massa da
partícula. [η] é expressa em unidade de volume por unidade de massa e está directamente relacionada
com o volume hidrodinâmico da partícula.
[η] depende:
- massa molecular
- da interacção entre os segmentos do polímero e das moléculas do solvente
Quanto maior for a interacção, tanto maior (mais inchado) será o novelo do polímero.
↑interacção→↑resistência→↑viscosidade
Quando as interacções entre a partícula e o solvente são mínimas ou nulas, a partícula adquire uma
morfologia aproximadamente de uma esfera rígida. O volume hidrodinâmico da partícula é mínimo.
Para partículas esféricas (segundo Einstein)
A fracção de volume é também característico visto que depende apenas da viscosidade.
Modificação da equação por Simha para ter em conta partículas com outras formas (que não
esférica)](https://image.slidesharecdn.com/sebentafisicaaplicda-230208235124-4cc330c5/85/sebenta-FisicaApliCada-pdf-97-320.jpg)

















































![159
Determinação da pureza óptica
A partir dos valores obtidos com o polarímetro, pode-se verificar qual o grau de pureza da
substância.
1• Pureza óptica = excesso de enantiómero (%)
2• Pureza óptica = % enantiómero em excesso = % enantiómero 1 - % enantiómero 2
3• Pureza óptica = 100 [α] mistura / [α] amostra pura
Variáveis que afectam a rotação óptica
Só em condições padronizadas se podem comparar substâncias opticamente activas. Isto significa
que há factores que alteram a sua actividade óptica.
1- Comprimento de onda
O poder rotatório específico varia com o comprimento de onda de acordo com a função
Sendo:
K1; K2; K3 – constantes avaliadas experimentalmente
λ- Comprimento de onda da medida
λ1; λ2 – constantes aproximadamente identificadas com os comprimentos de onda máximos de
absorção](https://image.slidesharecdn.com/sebentafisicaaplicda-230208235124-4cc330c5/85/sebenta-FisicaApliCada-pdf-159-320.jpg)