Transferir como PDF, PPTX





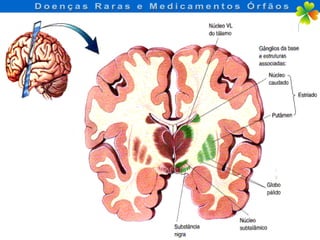
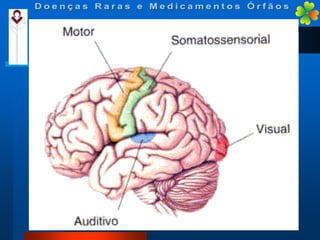
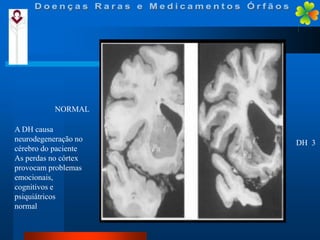
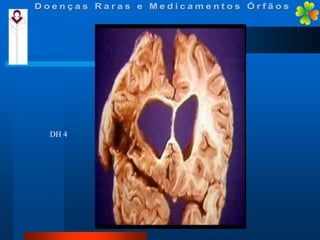


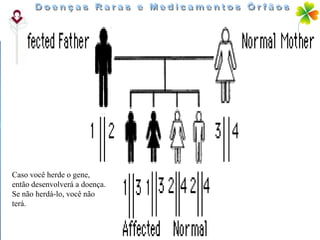































A Doença de Huntington (DH) é uma condição hereditária neurodegenerativa que causa deterioração cognitiva, motora e emocional, com sintomas geralmente surgindo entre os 30 e 50 anos. A doença é causada por uma alteração genética autossômica dominante, resultando em manifestações como coréia, distúrbios psiquiátricos e demência, afetando significativamente a qualidade de vida. O diagnóstico envolve testes clínicos e genéticos, enquanto o tratamento é sintomático, sem cura disponível.
































































































