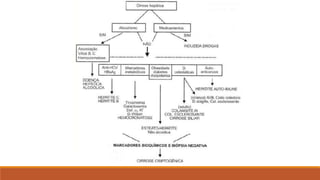
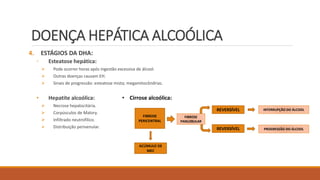
O documento resume as principais informações sobre cirrose hepática, incluindo:
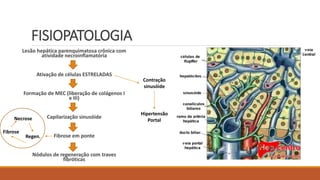
1) Definição, fisiopatologia e estágios da cirrose hepática;
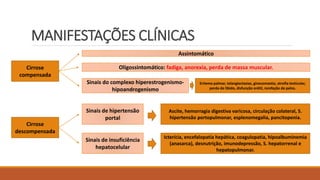
2) Manifestações clínicas da cirrose compensada e descompensada;
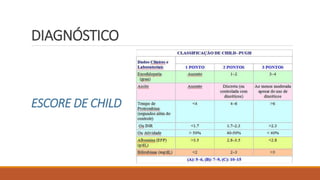
3) Diagnóstico da cirrose através de exames laboratoriais, de imagem e escore de Child;
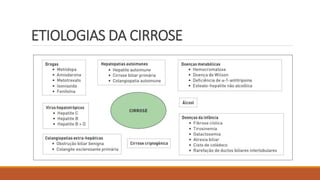
4) Principais causas da cirrose como doença hepática alcoólica, hepatites virais e esteatose hepática não alcoólica.