Transferir como PDF, PPTX




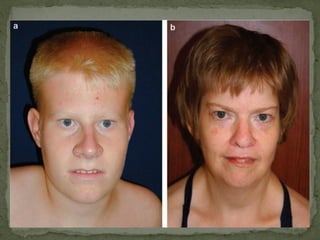





















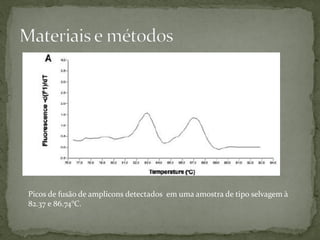
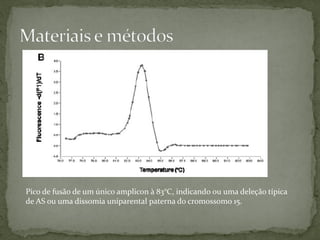
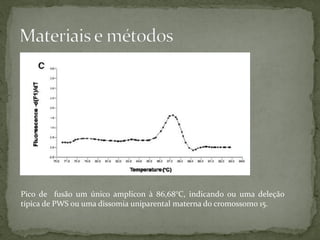












O documento descreve métodos para detectar a síndrome de Prader-Willi e Angelman analisando o padrão de metilação no gene SNRPN no cromossomo 15. A PCR específica de metilação e a análise de fusão de amplicons podem identificar deleções e dissomias uniparentais, enquanto a MS-MLPA permite detectar duplicações raras.

























































