Transferir como PDF, PPTX














![Referências Bibliográficas
• 1. Online Mendelian Inheritance in Man, OMIM (TM) [online]. Baltimore: Johns Hopkins
University; 2001. [cited 2001 Nov 12]. Avaliable from:URL:
http://www.ncbi.nlm.nih.gov/omim/.
• 2. Marfan’s syndrome: an overview. Yuan S-M, Jing H. Sao Paulo Med J. 2010;128(6):360-6.
• 3. Robinson PN, Godfrey M. The molecular genetics of Marfan syndrome and related
microfibrillopathies. J Med Genet. 2000;37(1):9-25.
• 4. Fibrillin Mutations. Do they really cause Marfan syndrome? Available from:
http://www.ctds.info/fibrillin.html. Accessed in 2010 (Set 29).
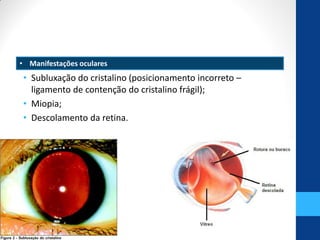
• 5. Anomalias oculares e características genéticas na síndrome de Marfan. Sallum, J. M. F. et
al. Arq Bras Oftalmol 2002;65:623-8.
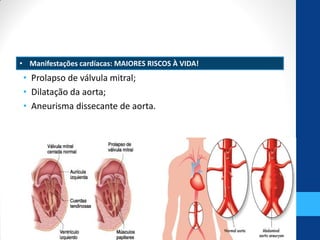
• 6. Tahernia AC. Cardiovascular anomalies in Marfan’s syndrome: the role of
echocardiography and beta-blockers. South Med J. 1993;86(3):305-10.
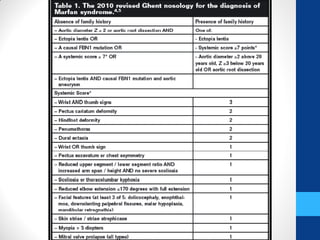
• 7. http://www.marfan.org/dx/revised-ghent-nosology.
• 8. http://www.marfan.com.br/index.asp.
• 9. http://www.marfan.org/about.
• 10. Dietz HC, Loeys B, Carta L, Ramirez F. Recent progress towards a molecular
understanding of Marfan syndrome. Am J Med Genet C Semin Med Genet. 2005;139C(1):4–
9.
• 11. PEDIATRICS Volume 132, Number 4, October 2013. FROM THE AMERICAN ACADEMY OF
PEDIATRICS.](https://image.slidesharecdn.com/slidessndromedemarfan-150113084036-conversion-gate01/85/Sindrome-de-Marfan-20-320.jpg)


A Síndrome de Marfan é uma doença genética do tecido conjuntivo causada por mutações no gene FBN1. Manifesta-se principalmente no sistema esquelético, cardiovascular e ocular, com riscos à vida como dilatação da aorta. O diagnóstico é clínico com base nos critérios de Ghent atualizados em 2010 e o tratamento envolve controle multidisciplinar e cirurgia profilática para prevenir complicações.


































































