Ailton Mascarenhas doSantos Filho, Beatriz Rivolt Trindade, Camila
Cason Narciso, Laura Penna Marins, Lucas Braga Manzatto e Maria
Helena Madalosso Senger
Doenças hereditárias do
metabolismo do glicogênio
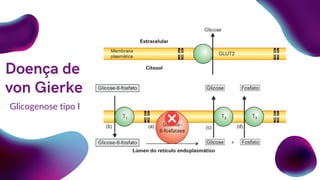
Doença de von Gierke
Doença de Pompe
Doença de Cori
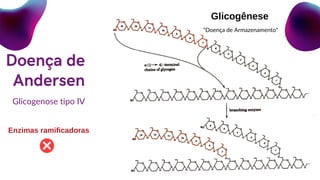
Doença de Andersen
Doença de McArdle
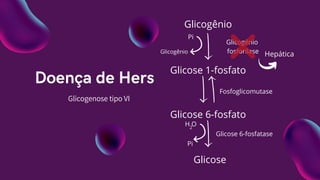
Doença de Hers
Glicogenoses
Aspectos Gerais eConsequências
Desordem metabólica genética
autossômica recessiva.
Cerca de 25% das glicogenoses.
Se manifesta nos primeiros meses de
vida.
Ausência parcial ou total da enzima
Glicose6-Fosfatase.
Acúmulo do glicogênio hepático.
Hipoglicemia.
Hiperlipidemia.
Acidose Lática.
Hiperuricemia --> Gota.
4.
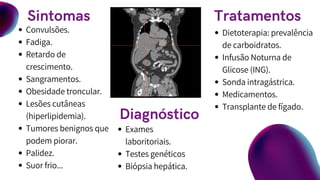
Convulsões.
Fadiga.
Retardo de
crescimento.
Sangramentos.
Obesidade troncular.
Lesõescutâneas
(hiperlipidemia).
Tumores benignos que
podem piorar.
Palidez.
Suor frio...
Sintomas
Diagnóstico
Tratamentos
Exames
laboritoriais.
Testes genéticos
Biópsia hepática.
Dietoterapia: prevalência
de carboidratos.
Infusão Noturna de
Glicose (ING).
Sonda intragástrica.
Medicamentos.
Transplante de fígado.
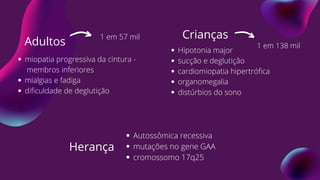
Crianças
Hipotonia major
sucção edeglutição
cardiomiopatia hipertrófica
organomegalia
distúrbios do sono
Adultos
miopatia progressiva da cintura -
membros inferiores
mialgias e fadiga
dificuldade de deglutição
Herança
Autossômica recessiva
mutações no gene GAA
cromossomo 17q25
1 em 57 mil
1 em 138 mil
7.
Diagnóstico
Biópsia muscular oude
fibroblastos da pele
Genotipagem a partir de
cultura sanguínea
Tratamento
sintomático
substituição enzimática:
alfa-alglucosidase
recombinante humana
Eficaz
Prognóstico depende do
estágio da doença.
Rara autossômica recessiva
Mutaçõesno gene AGL (1p21)
Há resposta ao glucagon
Não há maiores taxas de ácido
láctico
Sintomas característicos
Testes genético-moleculares
Aspectos gerais Tipos
IIIa:
~ 85% dos pacientes;
Fígado e músculo
IIIb:
~ 15% dos pacientes
Apenas fígado
Diagnóstico
10.
Sintomas
Hepatomegalia
Hipoglicemia cetótica comjejum
Concentrações elevadas de
transaminases e CK
Fraqueza muscular (IIIA)
Infância: hiperlipidemia
Adolescência: menos sintomas
hepáticos;
Cardiomiopatia hipertrófica e miopatia
esquelética (IIIa)
Tratamentos
Dieta hiperprotéica
Bebês: amamentação
mais recorrente
Transplante hepático
(casos graves)
Prevenção de sintomas:
bom controle
metabólico
Apresentações são ferramentas
decomunicação que podem
ser usadas como
demonstrações, palestras,
discursos, relatórios e mais. É
apresentada principalmente
diante de um público.
02
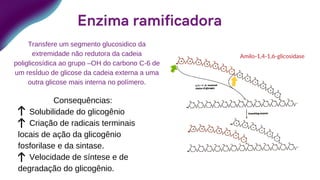
Transfere um segmento glucosidico da
extremidade não redutora da cadeia
poliglicosídica ao grupo –OH do carbono C-6 de
um resÍduo de glicose da cadeia externa a uma
outra glicose mais interna no polímero.
Consequências:
Solubilidade do glicogênio
Criação de radicais terminais
locais de ação da glicogênio
fosforilase e da sintase.
Velocidade de síntese e de
degradação do glicogênio.
Enzima ramificadora
Amilo-1,4-1,6-glicosidase
13.
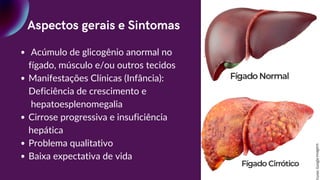
Aspectos gerais eSintomas
Acúmulo de glicogênio anormal no
fígado, músculo e/ou outros tecidos
Manifestações Clínicas (Infância):
Deficiência de crescimento e
hepatoesplenomegalia
Cirrose progressiva e insuficiência
hepática
Problema qualitativo
Baixa expectativa de vida
Font
e:
Goo
gle
Imag
ens
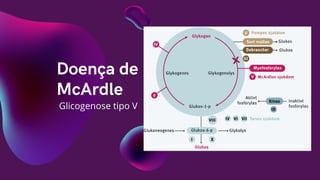
Descoberta por BrianMcardle em
1951
Acomete de 5 a 10 indivíduos por
milhão
Autossômica recessiva
Manifesta-se entre a
adolescência e ínicio da fase
adulta
Caracterizada pelo acúmulo de
glicogênio muscular
Falta de glicogênio fosforilase
muscular
Corpo passa a usar fosfatos e
gordura como fonte de energia
Trabalho muscular intenso pode
causar crise energética
Aspectos gerais
Sintomas
Dor muscular
Câimbras
Crises de mioglobinúria
17.
Tratamentos
Ingestão programada
de glicose
Exercíciosfísicos
controlados
Rabdomiólise
Insuficiência renal aguda
Hiperuricemia
Isquemia muscular
Gota
Cálculos renais
Complicações
Diagnóstico
Feita por um ortopedista
Exame de sangue - Creatina
quinase
Aspectos Gerais
Forma benigna
Doençaautossômica recessiva
Mutações no gene PYGL (14q21-q22)
Órgão atingido: Fígado
Doença de armazenamento de glicogénio do tipo 6b
Déficit da enzima glicogênio fosforilase hepática
Manifesta-se na infância
Causa atraso no crescimento, hepatomegalia
(desaparece na puberdade) e hipoglicemia leve.
20.
Sintomas frequentes
Controle eTratamento
Dieta com alta ingestão de
carboidratos
Maioria não precisa de
tratamento específico
Hepatomegalia
Aumento do conteúdo de
glicogênio hepático
Puberdade retardada
Transaminase hepática
elevada
Cetose
Osteopenia e Osteoporose
Diagnóstico
Revelação de glicogénio em
excesso e deficiência parcial
da fosforilase total e ativa na
biópsia hepática.
21.
Referências Bibliográficas
ADAM, MargaretP. et al. Gene Reviews. 1993 - 2021. Universidade de Washigton, Seatlle (EUA). "Glycogen Storage Disease Type III". Disponível em: <
https://www.ncbi.nlm.nih.gov/books/NBK26372/ >
TAKAHASHI, Hiroko Luzia et al. Determinação da atividade da fosforilase do glicogênio para o diagnóstico de doenças de estocagem do glicogênio.
Rev. bras. anal. clin, p. 63-68, 1999.
GUIMARÃES, Joana Maria Machado Mesquita. Terapêutica nutricional/alimentar nas doenças hereditárias do metabolismo dos hidratos de
carbono. 2021.
Portal das doenças raras e os medicamentos órfãos. Disponível em: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?
lng=PT&data_id=18&Disease_Disease_Search_diseaseGroup=Glicogenose-tipo-6-
&Disease_Disease_Search_diseaseType=Pat&Grupo%20de%20doen%E7as%20relacionadas=Glicogenose-tipo-6B--por-deficiencia-de-fosforilase-
hep-tica&title=Glicogenose%20tipo%206B,%20por%20defici%EAncia%20de%20fosforilase%20hep%E1tica&search=Disease_Search_Simple.
Acesso em 17 de nov. de 2021.
Socialstylerelsen. McArdles sjukdom. Disponível em: https://www.socialstyrelsen.se/stod-i-arbetet/sallsynta-halsotillstand/mcardles-sjukdom/.
Acesso em 03 de nov. de 2021
RAMARAL, V. F. M.; MARTINS, A. A. de S. Quando a preguiça é sinônimo de doença - um caso de doença de McArdle evista Brasileira de Medicina de
Família e Comunidade. 2016
Nunes FHS. Doença de Von Gierke: estudo de revisão. - Revista de Pediatria SOPERJ. 2009;10(1):21-27
Julia Maria Avelino Ballavenuto; Jéssica D´Ório Dantas de Oliveira; Renato Jorge Alves. Glicogenose Tipo I (Doença de Von Gierke): Relato de Dois
Casos com Grave Dislipidemia. Arq. Bras. Cardiol. vol.114 no.4 supl.1 São Paulo abr. 2020