


![Genoma
• Sequência(s) completa(s) de DNA
[cromossomo(s)] de um organismo específico –
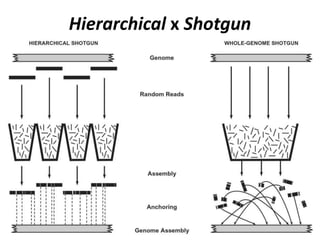
indivíduo – ou representantes [genoma
referência] para uma determinada espécie.
– Conjunto de todos os genes](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-3-320.jpg)

![Conceito de Gene
• 1866 - Conceito clássico em genética - Gregor Mendel
• Unidade discreta de herança ("fatores" heredítários);
• 1909 - O termo gene foi cunhado - Wilhelm L. Johannsen
• Conceito abstrato das unidades de herança ("entidade quasi mítica" [Keller, E.F, 2000]);
• 1915 - Teoria acerca dos cromossomos - Thomas Hunt Morgan
• Determinado locus em um cromossomo;
• 1941 - Conceito "um-gene-uma-enzima" - George W. Beadle e Edward L. Tatum
• 1953 - O gene começa a ganhar uma definição ainda mais materialista.
• Sequências de nucleotídeos;
• 1959 - Conceito "um-gene-um-polipeptídeo" - George W. Beadle e Edward L. Tatum
• 1961 - Conceitos de genes estruturais e regulatórios - François Jacob e Jacques Monod
• 1977 - Um-gene-múltiplos-produtos - Richard J. Robets e Phillip A. Sharp
• 1990 - Composição de domínios no DNA (Modelos da estrutura gênica) - Thomas Fogle;
• 1999 - Conceitos de "gene molecular" e "gene evolutivo" - Paul E. Griffiths; Eva M. Neumann-Held;
• …
• Conceito moderno: Entidade codificada em ácidos polinucléicos a qual ao menos pode ser transcrita
[Stephen T. Abedon].
Abstrato
Concreto](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-5-320.jpg)

![Definição operacional de gene
(proposta)
Gene é a união de sequências genômicas que
codificam um conjunto coerente de produtos funcionais
que potencialmente possuem regiões sobrepostas.
[Gerstein et al., 2007]
Segmentos de DNA
codificadores
de proteínas
(ORFs):
A
B
C
D
E](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-7-320.jpg)
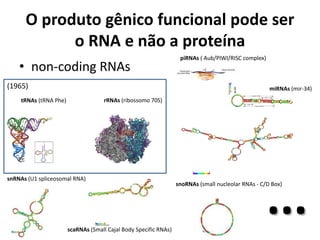
![Aspectos que devem ser considerados
• Gene é a união de sequências genômicas que
codificam um conjunto coerente de produtos
funcionais (que possuem regiões que se sobrepoem
considerando a referência genômica)
[Gerstein et al., 2007]
– Três aspectos devem ser considerados:
• Gene é uma sequência genômica que codifica precisamente um
produto funcional (RNA ou proteína);
• Nos casos onde há muitos produtos funcionais compartillhando
regiões sobrepostas, a união de todas as regiões de sequências
genômicas sobrepostas que codificam cada produto é um gene;
• A união deve ser coerente (RNA/proteína) – porém não requer
que todos os produtos necessariamente compartilhem
exatamente as mesmas regiões.](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-9-320.jpg)
![Dogma Central da Biologia Molecular
Crick F. Central dogma of molecular biology. Nature. 1970 Aug 8;227(5258):561-3. PubMed PMID: 4913914.
[Crick, F. , 1970]
transcrição
FLUXO DA INFORMAÇÃO GÊNICA
tradução
replicação
casos especiais](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-10-320.jpg)
![GENÔMICA ESTRUTURAL
Introdução
[Crick, F. , 1970]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-11-320.jpg)



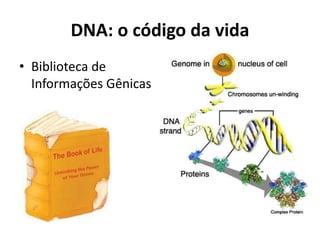


![Primeiros passos...
• Primeiros Métodos de Sequenciamento
• Baseados em eletroforese
WalterGilbert
FrederickSanger
Prêmio Nobel em Química - 1980
Método Químico
Tratamento químico para degradar o
DNA em nucleotídeos específicos para
posterior leitura
[Maxam e Gilbert, 1977]
Método Enzimático
Baseado na síntese enzimática de uma
fita complementar interrompida pela
incorporação de um didesoxinucleotídeo
(terminação da cadeia) para posterior leitura
[Sanger et al., 1977]
Prêmio Nobel em Química – 1958
Prêmio Nobel em Química – 1980](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-18-320.jpg)
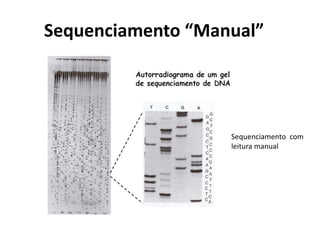











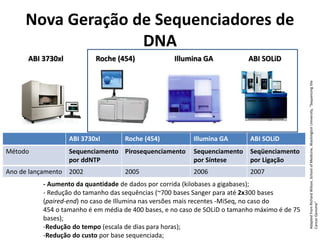








![Estratégia de sequenciamento
shotgun
[Commins, Toft e Fares, 2009]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-42-320.jpg)
![Mapeamento de leituras e
Montagem “de novo”
[Haas and Zody, Nature Biotechnology 28, 421–423 (2010)]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-43-320.jpg)

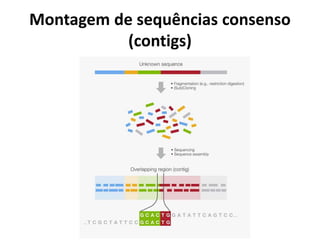
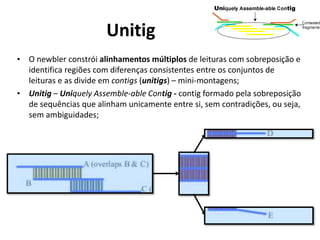
![Montagem “de novo”
de sequências
• Sequenciamento “de novo”
– Alinhamentos múltiplos de
sequências de leituras
(evidências experimentais)
• Montagem de fragmentos de
sequências genômicas originais
através de um consenso
CTGTTATGGGCAACCCTAAGGTGAAGGCTCATGGCAAGAAAGTGCTCGGTGCCTTTAGTGATGGCCTGGCTCACCTGTGGAC
ATGGGCAACCCTAAGGTGAAGGCT TGCTCGGTGCCTTTAGTGATGGCCTGGCTCACCTGTG
TAAATGGGCAACCCTAAG
GCCGGCAACCCGAAGGTG
CCTAAGGTGAAGGCTAGC
GTTTGCTCGGTGCCTTTA
GTGCCTTTAGTGATGAAA
GATGGCCTGGCTCACAGC
GCCCCTGGCTCACCTGTG
Original:
Consensus :
Read 1
Read 2
Read 3
Read 4
Read 5
Read 6
Read 7
[Blanca, J. COMAV Institute]
Original:
Consensus :
Leitura 1
Leitura 2
Leitura 3
Leitura 4
Leitura 5
Leitura 6
Leitura 7
Leitura 8
Leitura 9
Leitura 10
Leitura 11
ACCCTAAGGTGAAGG
CCGAAGGTGAAGGCT
GGCAACCCTAAGGTG
GCAACCCGAAGGTGA
ATGGGCAACCCGAAGGTGAAGGCT](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-45-320.jpg)





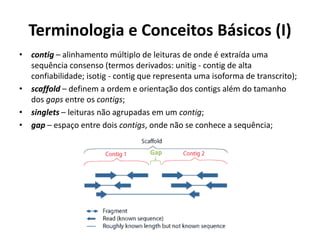
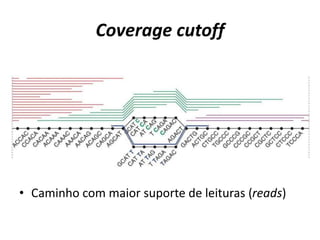
![Terminologia e Conceitos Básicos (II)
• Cobertura (coverage) – fold coverage
– Total de bases sequenciadas [N * L] dividido pelo
tamanho da região de interesse (e.g. genoma) [G]
• (N * L)/G
– N = Número de leituras
– L = Tamanho da leitura
– G = Tamanho da região de interesse
• Exemplo
– Tamanho do Genoma (G): 1 Mbp
– Quantidade de leituras (N): 5 milhões de reads
– Tamanho das leituras (L): 50 bp
» Cobertura = (5.000.000 * 50) / 1.000.000 = 25X
– Na prática, corresponde a quantas vezes, em média,
cada base do alvo (genoma) foi sequenciada;](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-53-320.jpg)
![Terminologia e Conceitos Básicos (III)
• Cobertura necessária em projetos de
sequenciamento de genomas:
– Resequenciamento:
• Sanger (Leituras de ~800bp): C. Venter (3Gb ~7.5x)
– [Levy et al., 2007]
• Roche 454 (Leituras de ~400bp): J. Watson (3Gb ~7.4x)
– [Wheeler et al., 2008]
– Sequenciamento “de novo”:
• Illumina (Leituras de 52pb): Panda (Ailuropoda
melanoleura) (2,4Gb ~56x)
– [Li et al., 2010]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-54-320.jpg)
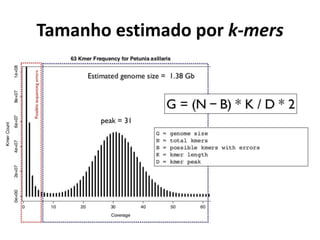
![Como estimar os parâmetros de
sequenciamento?
Estimar parâmetros (número esperado de contigs, tamanho dos contigs)
[Lander e Waterman, 1988]
Considerações:
Amostragem equivalente a um processo de Poisson;
Assume que as leituras serão amostradas aleatoriamente no genoma;
L = tamanho das leituras
T = mínimo de sobreposição entre as leituras
G = tamanho do genoma
N = número de leituras
c = cobertura = (N*L/G)
σ = 1 –(T/L)
e = 2,718
E(número de contigs) = Ne(-c*σ)
E(tamanho dos contigs) = L*( ((e(c*σ)–1)/c) + (1–σ) )
Modelo Lander-Waterman](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-55-320.jpg)
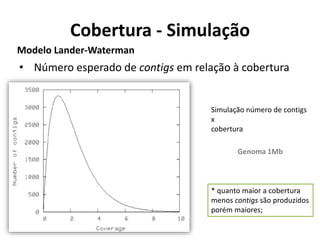

![Será que o modelo se aplica aos
dados de NGS?
• Genoma do Panda (Ailuropoda melanoleura)
– Tamanho do genoma 2,4 Gb
C = (N*L)/G C = 8x
G = 2.400.000.000 (2,4Gb)
L = 52 pb
[Li R et al., 2010]
8 = (N*52)/2400000000
52*N = 8*2400000000
N=19200000000/52
N=369.230.769
37 bibliotecas do tipo paired-end e mate-pair (150 bp, 500 bp, 2 kbp, 5 kbp, and 10 kbp)
Média de tamanho de 52 pb
218 lanes Illumina Genome Analyzer (17 lanes descartadas por baixa qualidade)
3.379.000.000 de reads (96% cobertura do genoma)
176 Gb (73×) de cobertura – fold coverage (reads utilizáveis)
134 Gb (56×) de cobertura – fold coverage (reads de alta qualidade)](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-58-320.jpg)


![Cobertura – nova geração de
sequenciadores
• Tamanho esperado de contigs em relação à cobertura
Panda e Cachorro
genomas de ~2,4Gb[Schatz et al., 2010]
Discrepância grande
entre o predito (Modelo LW) e o
observado (média e N50)
Resultado de um modelo
simplificado, que não leva em
consideração:
- leituras curtas e genomas
repetitivos;
- qualidade das leituras;
- sequenciamento não uniforme
-vieses (ex.: conteúdo de GC);
- ...](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-61-320.jpg)
![Importância do tamanho das leituras
[Whiteford et al., 2005]
Contigs > que o tamanho
indicado no gráfico.
200000 = ~35% genoma
de E. coli
Leituras de tamanho 200](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-62-320.jpg)
![Tamanho do Genoma
• Quantidade total de DNA contido dentro de
um genoma (cópia única – genoma haplóide).
– Valor C [ C-value ]
• Massa
– Picograma (trilionésimo [10-12] de grama - pg)
• Número total de nucleotídeos em pares de
base (pb)
– 1 pg = 978 pb](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-63-320.jpg)





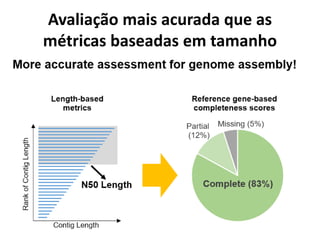
![N = tamanho ?
L = quantidade ?
“(…)We used a statistic called the ‘N50 length’, defined as the largest length L
such that 50% of all nucleotides are contained in contigs of size at least L. (…)”
http://www.acgt.me/blog/2015/6/11/l50-vs-n50-thats-another-fine-mess-that-
bioinformatics-got-us-into
[International Human Genome Sequencing Consortium Lander et al., 2001]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-69-320.jpg)



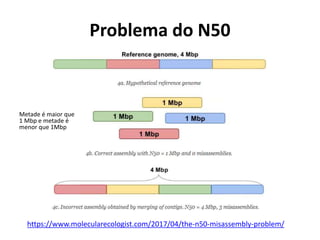
![Uma outra métrica…
• NG50
[Considera o tamanho estimado do genoma para definir o que está acima de 50%]
http://www.molecularecologist.com/2017/04/a-solution-to-the-n50-filtering-problem/
NG50 = o cálculo do N50 é realizado com o valor estimado
do tamanho do genoma. Neste caso 500 kbp e não com o
tamanho da montagem 400 kbp.](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-74-320.jpg)
![outra métrica…
• NA50
– N50 para o conjunto
de blocos alinhados
(ao invés do
conjunto inicial de
contigs)
• Dessa forma, se
alguns dos contigs
falham em alinhar,a
métrica NA50 ainda
é computada com
respeito aos 50% da
montage total
(incluindo os contigs
que alinharam e
não alinharam)
Necessita haver uma sequência referência onde os blocos podem alinhar
https://www.molecularecologist.com/2017/04/a-solution-to-the-n50-misassembly-problem/
[Considera o tamanho da montagem para os 50%]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-75-320.jpg)



![COMPASS
[https://gigascience.biomedcentral.com/articles/10.1186/2047-217X-2-10.ris]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-79-320.jpg)

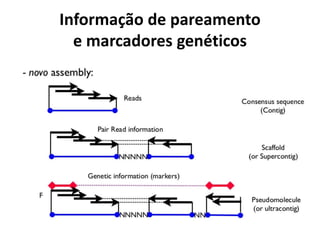
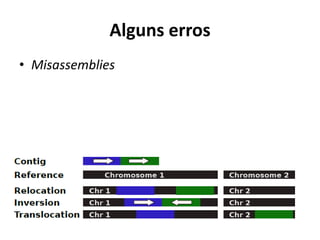

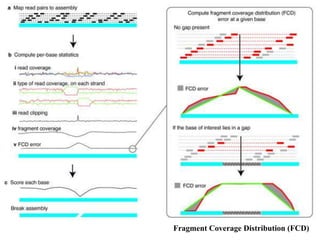
![QUAST
• MISASSEMBLIES
– No. of misassemblies:
• Número de erros, usando definição de Plantagora [Barthelson et al., 2011]
– misassembly breakpoint : posição no contig onde a sequência no flanco esquerdo
com relação à sequência no flanco direito neste alinhamento com a referência
» alinha acima de 1kb de distância;
» sobrepõem acima 1 Kb;
» alinha em fitas opostas;
» alinha em diferentes cromossomos;
– No. of misassembled contigs:
• Número de contigs que contêm misassembly breakpoints.
– Misassembled contigs length:
• Número de bases em todos os contigs com um ou mais misassemblies.
– No. of unaligned contigs:
• Número de contigs que não têm alinhamento com a sequência referência.
– No. of ambiguously mapped contigs:
• Número de contigs que têm bom mapeamento (altos escores e idênticos) em múltiplos
locais no genoma.
QUAST também provê relatório com detalhamento dos contigs que estão em cada categoria.](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-85-320.jpg)

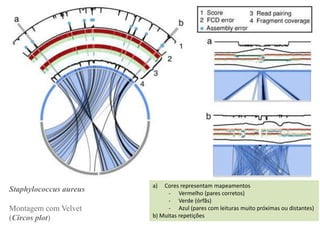



![Generalizado e cópia simples
Dissecting the Drosophila melanogaster gene set by orthologous group universality and duplicability highlights how the largest
fractions of genes are preserved as single-copy orthologues across all 80 insects or specific to the 12 drosophilids. Orthologous
groups with 80 insect species from OrthoDB: universality, from widespread to specific or sparse species representation; duplicability,
from mostly single-copy to mostly multi-copy orthologue counts.
[https://doi.org/10.1016/j.cois.2015.01.004]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-93-320.jpg)
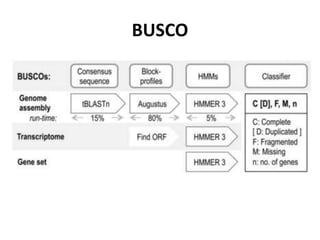

![gVolante
• https://gvolante.riken.jp/
Comparação com avaliações [CEGMA, CVG, BUSCO] de genomas pré-computadas.
[https://doi.org/10.1093/bioinformatics/btx445]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-96-320.jpg)


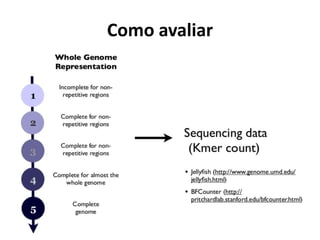






![Viés na composição
[Hansenetal.,2010]
Mapeamento genômico
DOI: 10.1093/nar/gkq224
[Hansen et al., 2010]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-107-320.jpg)
![Viés no conteúdo de GC (1)
Sequenciamento do Panda Gigante
(Ailuropoda melanoleuca)
[Li R et al., 2010] [Li R et al., 2010]
% GENOMA MONTADO x CONTEÚDO G+C QUANTIDADE DE LEITURAS x CONTEÚDO G+C](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-108-320.jpg)
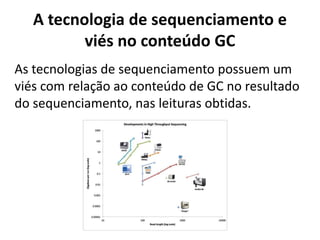
![Viés no conteúdo de GC
nas plataformas NGS
[Quail et al., 2012]Viés: Illumina livre de amplificação [Kozarewa I, et al., 2009] (menor o viés)
/ PGM (maior o viés)
Genoma protozoário
Plasmodium falciparum (19,4%GC)](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-110-320.jpg)
![Erros inerentes às plataformas de
sequenciamento
[Fox et al., 2014]
doi:10.4172/jngsa.1000106](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-111-320.jpg)
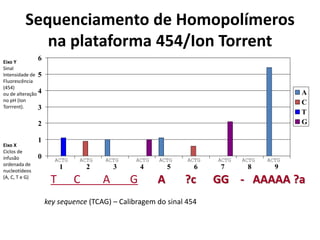
![Erros no Sequenciamento de
Homopolímeros na plataforma 454
Linearidade mantida até
homopolímeros de tamanho 8 nt
Distribuição dos erros
em homopolímeros
[Margulies M, et al. , 2006]
[Margulies M, et al. , 2006]
Dentre os erros até 6-mers:
Inserções (azul) Deleções (vermelho)](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-113-320.jpg)


![k-mers Uniqueness ratio
k-mers uniqueness ratio – número de k-mers distintas que ocorrem uma única vez no genoma
número total de k-mers distintas que ocorrem no genoma
[Schatz et al., 2010]
Trichomonas vaginalis
Exige um tamanho maior
de k-mer para alcançar a
unicidade](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-116-320.jpg)

![Rochas, pedras e pedregulhos
[http://www.genomenewsnetwork.org/articles/03_00/assemble_genome_3_24.shtml]
paired-end/mate-pair
Classificação desses fragmentos
nos montadores:
mais confiáveis (Rochas) aos
menos confiáveis (Pedregulhos)](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-118-320.jpg)




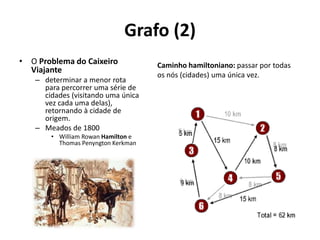
![Grafo (1)
• Grafo é uma estrutura abstrata, pode ser representada por uma
rede de nós conectados por arcos
• Königsberg (Kaliningrad, Russia)
– Século 18
• Problema proposto: Cruzar as sete pontes numa caminhada contínua sem
passar duas vezes por qualquer uma delas.
• Caminho euleriano: passar por todas as arestas (pontes) uma única vez.
• Solução: não há (nós possuem valência – ou grau – ímpar)
Regiões de Königsberg = nós ou vértices
Pontes = arcos ou arestas[Compeau, Pevzner & Tesler, 2011]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-123-320.jpg)
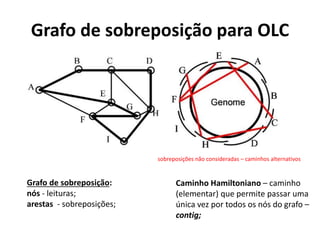
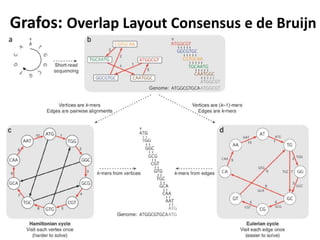
![Overlap-Layout-Consensus (OLC)
• 1º detecção de sobreposição;
– Alinhamento pareado entre todas as
leituras – identificação dos pares com
melhor match (alinhamento
global/local + heurísticas [e.g. seed &
extend]);
• 2º layout dos fragmentos (montagem do
contig);
– Construção e manipulação do grafo de
sobreposição
(Analisar/Simplificar/Limpar);
– Caminho Hamiltoniano;
• 3º decisão da sequência (montagem do
consenso);
– Alinhamento Múltiplo de Sequências
(Layout obtido percorrendo o(s)
caminho(s) mais provável(l/is) –
maior suporte);
– Obtenção da sequência consenso
(Normalmente a frequência de um
nucleotídeo em determinada
posição determina a base
consenso;)](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-125-320.jpg)

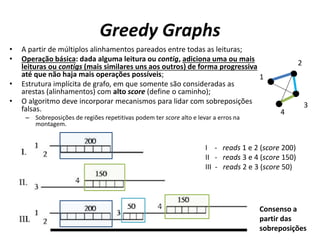


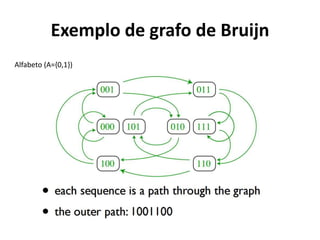
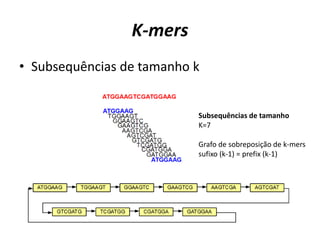

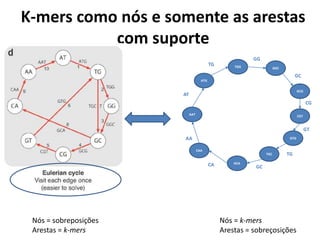
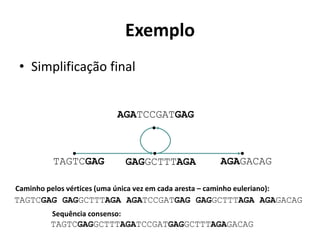
![Estratégia utilizando grafos de-Bruijn
[Schatz M C et al. Genome Res. 2010;20:1165-1173]
Grafo de k-mers (subsequências de tamanho k = 3)
e sobreposições de tamanho k-1 (3-1=2)
Grafo de Reads e
suas sobreposições](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-137-320.jpg)


![Tamanho de k
• Tamanho de k :não pode ser nem muito grande, nem muito pequeno:
– grande o suficiente para não pegar falsas sobreposições que
compartilham k-mers em comum (resolução de repetições);
• k-mers grandes
– menor conectividade nos grafos com maior especificidade;
– grafos menores consomem menos memória RAM;
– pequeno o suficiente para encontrar o máximo de sobreposições
verdadeiras (maior aproveitamento, lidando com pequenos erros de
sequenciamento);
• k-mers pequenos
– alta conectividade nos grafos com maior sensibilidade;
– maior divergência e ambiguidade;
– grafos maiores consomem mais memória RAM;
• Solução para minimizar o problema:
– Combinar as informações dos grafos com diferentes tamanhos de k;
• [Surget-Groba et al., 2010]
• [Schulz et al., 2012]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-140-320.jpg)

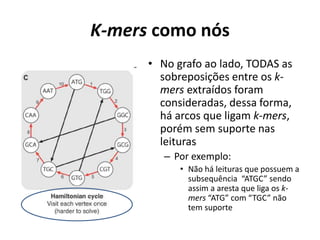
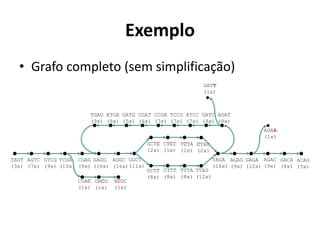
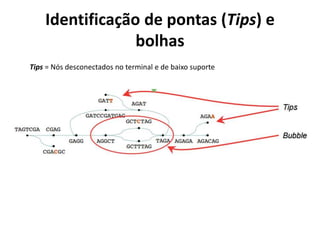
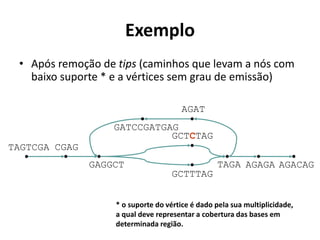
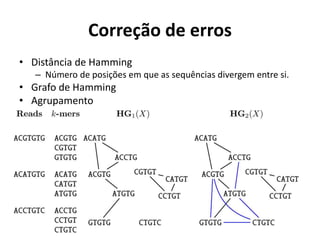
![Complexidades em k-mers
• Ramificações – caminhos sem-saídas
divergentes;
– Induzidos por erros no sequenciamento nas
extremidades das leituras;
• Bolhas – caminhos que divergem e depois
convergem;
– Induzidos por erros no sequenciamento no meio
das leituras;
• Corda esfiapada – caminhos que convergem e
divergem;
– Induzidos por repetições;
• Ciclos – caminhos que convergem neles
mesmos;
– Induzidos por repetições (e.g. repetições em
tandem – pequenos ciclos);
[Miller, J.R., et al., 2010]
"tips"](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-142-320.jpg)





![SPAdes x Velvet
• 50 Salmonella enterica subsp. enterica serovar Paratyphi B dTa+ (S. Java) isolates were tested. DNA
[http://www.engage-europe.eu/-/media/Sites/engage-europe/Final-website-documents/ENGAGE_AppE_benchmarking_Velvet-
SPAdes_final.ashx?la=da&hash=A6AB88A45DC9205300258FCD824D4C7304214551]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-153-320.jpg)



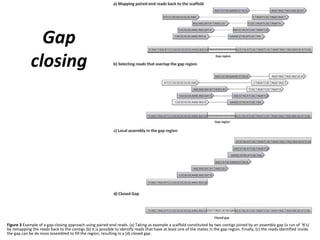
![Sobreposições
[http://www.genomenewsnetwork.org/articles/03_00/assemble_genome_3_24.shtml]
ERRO DE MONTAGEM FRAGMENTO IGNORADO](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-158-320.jpg)
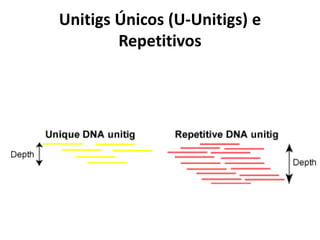


![Chamada básica do Montador
runAssembly [parâmetros] seqs.fasta
• Procura pelo arquivo seqs.fasta.qual no
mesmo diretório
• Cria o seguinte diretório (por padrão):
– P_yyyy_mm_dd_hh_min_sec_runAssembly
• P_ = Projeto, seguido de data e hora
2.6+ - aceita sequências no formato FASTQ](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-165-320.jpg)







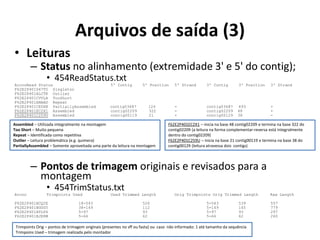

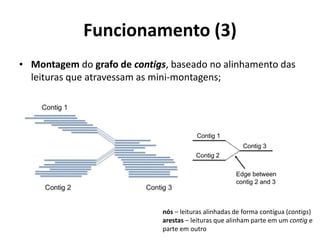
![Arquivos de saída (5)
• Grafos
– Estrutura de conexão entre contigs [3 seções – Nós (1) /Arestas (2)(3)];
• 454ContigGraph.txt
(1) ContigNum ContigName Length Average_depth
...
31 contig00031 12 1.4
32 contig00032 1633 80.3
33 contig00033 947 105.7
...
(2) Edge FromContigNum FromEnd ToContigNum ToEnd AlignmentReadDepth
...
C 32 5' 31 3' 5
C 32 3' 33 5' 20
...
S 22 2592 31:+;32:+;33:+
S 23 2580 32:+;33:+
S 24 947 33:+
...
(3) Edge ContigNum Sequence Thru-FlowInformation
...
I 4 TGTTCGGTGTTCTCCGCCTCGGGCTGTCACAAATCGTGCTGCTGTGAGCCACTGCGTGCAGGTCTCAT 2:2-3'..3-5';1:6-3'..3-5'
...
– Layout dos Isotigs
• 454IsotigsLayout.txt
>isogroup00007 numIsotigs=3 numContigs=3
Length : 12 1633 947 (bp)
Contig : 00031 00032 00033 Total:
isotig00022 >>>>> >>>>> >>>>> 2592
isotig00023 >>>>> >>>>> 2580
isotig00024 >>>>> 947
"I" short contig - seq. acima inicia antes do contig4 e termina depois = dois fluxos
de informação separados por ; qtd de sequências:contig_anterior-
extremidade..contig_posterior extremidade
"P" paired-ends – como as sequências em pares atravessam contigs e permitem
scaffolds
"F" read-flow – como as sequências simples atravessam contigs e permitem
scaffolds](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-175-320.jpg)

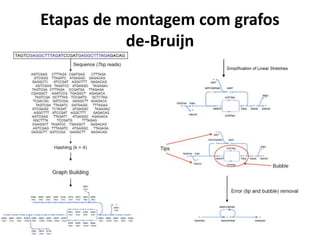
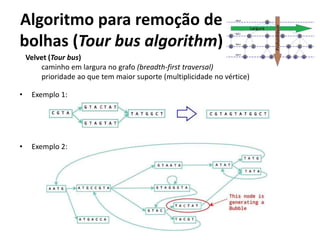
![Velvet: Pebble and Rock Band
• Resolução de Repetições e Scaffolding
– Paired-end sequencing (Pebble, Breadcrumb)
– Long-read sequencing (Rock Band)
Pebble
[Zerbino e Birney, 2009]
[ZerbinoeBirney,2009]
Rock Band
Breadcrumb
[ZerbinoeBirney,2008]
(miolo de pão)](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-178-320.jpg)
![Construção da tabela hash
• velveth
– Extração dos k-mers e indexação por meio de uma tabela hash a partir de um conjunto de
leituras. As sobreposições entre os k-mers imediatamente são obtidas.
– São gerados 2 arquivos (Sequences e Roadmaps) necessários para a construção do grafo de-
Bruijn pelo programa seguinte: velvetg;
• Sequences: sequências indexadas;
• Roadmaps: representação das sobreposições entre os k-mers únicos;
./velveth output_directory hash_length [[-file_format] [-read_type] filename]
• Principais parâmetros
– hash_length é o tamanho dos k-mers em bp. Quanto menor o k mais lento!!!
– read_type pode ser:
• -short / -shortPaired
• -short2 / -shortPaired2
• -long / -longPaired
– file_format pode ser:
• -fasta (default)
• -fastq
• ...
Hash Table (Array Associativo)
1 | ACGACA
2 | CGACAT
k-mer=3
ACG 1
CGA 1 2
GAC 1 2
ACA 1 2
CAT 2
K-mer](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-179-320.jpg)

![Construção do Grafo de-Bruijn (1)
• velvetg
– Construção e manipulação do grafo de-Bruijn, correção de erros e
resolução de repetições.
– Arquivos gerados:
• contigs.fa - sequências consensos (gaps dentro contigs = N's);
• PreGraph - grafo intermediário 0;
• Graph - grafo intermediário 1;
• Graph2 - grafo intermediário 2;
• LastGraph - descrição plena do grafo de-Bruijn produzido;
• Log - descrição das ações executadas;
• stats.txt - números relativos à montagem;
• UnusedReads.fa - sequências não utilizadas na montagem;
• velvet_asm.afg - formato compatível com AMOS (-amos_file yes);
./velvetg output_directory [options]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-181-320.jpg)





![VelvetOptimiser
• Encontrar os "melhores" parâmetros (k-mer e cov_cutoff)
– VelvetOptimiser.pl [options] -f 'velveth input line'
--help This help.
--v|verbose+ Verbose logging, includes all velvet output in the logfile. (default '0').
--s|hashs=i The starting (lower) hash value (default '19').
--e|hashe=i The end (higher) hash value (default '31').
--f|velvethfiles=s The file section of the velveth command line. (default '0').
--a|amosfile! Turn on velvet's read tracking and amos file output. (default '0').
--o|velvetgoptions=s Extra velvetg options to pass through. eg. -long_mult_cutoff -max_coverage etc (default '').
--t|threads=i The maximum number of simulataneous velvet instances to run. (default '48').
--g|genomesize=f The approximate size of the genome to be assembled in megabases.
Only used in memory use estimation. If not specified, memory use estimation
will not occur. If memory use is estimated, the results are shown and then program exits. (default '0').
--k|optFuncKmer=s The optimisation function used for k-mer choice. (default 'n50').
--c|optFuncCov=s The optimisation function used for cov_cutoff optimisation. (default 'Lbp').
--p|prefix=s The prefix for the output filenames, the default is the date and time in the format DD-MM-YYYY-HH-MM_.
(default 'auto').
Advanced!: Changing the optimisation function(s)
Velvet optimiser assembly optimisation function can be built from the following variables.
Lbp = The total number of base pairs in large contigs
Lcon = The number of large contigs
max = The length of the longest contig
n50 = The n50
ncon = The total number of contigs
tbp = The total number of basepairs in contigs
Examples are:
'Lbp' = Just the total basepairs in contigs longer than 1kb
'n50*Lcon' = The n50 times the number of long contigs.
'n50*Lcon/tbp+log(Lbp)' = The n50 times the number of long contigs divided
by the total bases in all contigs plus the log of the number of bases
in long contigs.](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-187-320.jpg)



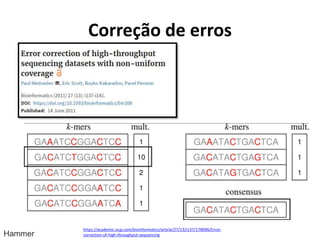
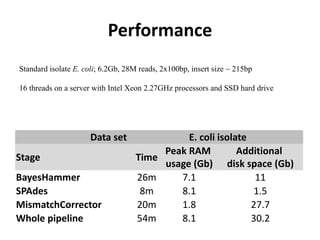
![SPAdes pipeline
• Módulos
– BayesHammer – read error correction tool for Illumina reads,
which works well on both single-cell and standard data sets.
– IonHammer – read error correction tool for IonTorrent data,
which also works on both types of data.
– SPAdes – iterative short-read genome assembly module; values
of K are selected automatically based on the read length and
data set type.
– MismatchCorrector – a tool which improves mismatch and
short indel rates in resulting contigs and scaffolds; this module
uses the BWA tool [Li H. and Durbin R., 2009];
MismatchCorrector is turned off by default, but we recommend
to turn it on (see SPAdes options section).](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-191-320.jpg)
![Linha de ComandoSPAdes genome assembler v3.13.0
Usage: /usr/local/bin/spades.py [options] -o <output_dir>
Basic options:
-o <output_dir> directory to store all the resulting files (required)
--sc this flag is required for MDA (single-cell) data
--meta this flag is required for metagenomic sample data
--rna this flag is required for RNA-Seq data
--plasmid runs plasmidSPAdes pipeline for plasmid detection
--iontorrent this flag is required for IonTorrent data
--test runs SPAdes on toy dataset
-h/--help prints this usage message
-v/--version prints version
Input data:
--12 <filename> file with interlaced forward and reverse paired-end reads
-1 <filename> file with forward paired-end reads
-2 <filename> file with reverse paired-end reads
-s <filename> file with unpaired reads
--merged <filename> file with merged forward and reverse paired-end reads
--pe<#>-12 <filename> file with interlaced reads for paired-end library number <#> (<#> = 1,2,...,9)
--pe<#>-1 <filename> file with forward reads for paired-end library number <#> (<#> = 1,2,...,9)
--pe<#>-2 <filename> file with reverse reads for paired-end library number <#> (<#> = 1,2,...,9)
--pe<#>-s <filename> file with unpaired reads for paired-end library number <#> (<#> = 1,2,...,9)
--pe<#>-m <filename> file with merged reads for paired-end library number <#> (<#> = 1,2,...,9)
--pe<#>-<or> orientation of reads for paired-end library number <#> (<#> = 1,2,...,9; <or> = fr, rf, ff)
--s<#> <filename> file with unpaired reads for single reads library number <#> (<#> = 1,2,...,9)
--mp<#>-12 <filename> file with interlaced reads for mate-pair library number <#> (<#> = 1,2,..,9)
--mp<#>-1 <filename> file with forward reads for mate-pair library number <#> (<#> = 1,2,..,9)
--mp<#>-2 <filename> file with reverse reads for mate-pair library number <#> (<#> = 1,2,..,9)
--mp<#>-s <filename> file with unpaired reads for mate-pair library number <#> (<#> = 1,2,..,9)
--mp<#>-<or> orientation of reads for mate-pair library number <#> (<#> = 1,2,..,9; <or> = fr, rf, ff)
--hqmp<#>-12 <filename> file with interlaced reads for high-quality mate-pair library number <#> (<#> = 1,2,..,9)
--hqmp<#>-1 <filename> file with forward reads for high-quality mate-pair library number <#> (<#> = 1,2,..,9)
--hqmp<#>-2 <filename> file with reverse reads for high-quality mate-pair library number <#> (<#> = 1,2,..,9)
--hqmp<#>-s <filename> file with unpaired reads for high-quality mate-pair library number <#> (<#> = 1,2,..,9)
--hqmp<#>-<or> orientation of reads for high-quality mate-pair library number <#> (<#> = 1,2,..,9; <or> = fr, rf, ff)
--nxmate<#>-1 <filename> file with forward reads for Lucigen NxMate library number <#> (<#> = 1,2,..,9)
--nxmate<#>-2 <filename> file with reverse reads for Lucigen NxMate library number <#> (<#> = 1,2,..,9)
--sanger <filename> file with Sanger reads
--pacbio <filename> file with PacBio reads
--nanopore <filename> file with Nanopore reads
--tslr <filename> file with TSLR-contigs
--trusted-contigs <filename> file with trusted contigs
--untrusted-contigs <filename> file with untrusted contigs](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-195-320.jpg)
![Linha de ComandoSPAdes genome assembler v3.13.0
Usage: /usr/local/bin/spades.py [options] -o <output_dir>
Basic options:
-o <output_dir> directory to store all the resulting files (required)
Input data:
-1 <filename>file with forward paired-end reads
-2 <filename>file with reverse paired-end reads
-s <filename>file with unpaired reads
--pe<#>-12 <filename> file with interlaced reads for paired-end library number <#> (<#> = 1,2,...,9)
--pe<#>-1 <filename> file with forward reads for paired-end library number <#> (<#> = 1,2,...,9)
--pe<#>-2 <filename> file with reverse reads for paired-end library number <#> (<#> = 1,2,...,9)
--pe<#>-s <filename> file with unpaired reads for paired-end library number <#> (<#> = 1,2,...,9)
--pe<#>-m <filename> file with merged reads for paired-end library number <#> (<#> = 1,2,...,9)
--pe<#>-<or> orientation of reads for paired-end library number <#> (<#> = 1,2,...,9; <or> = fr,
rf, ff)
--s<#> <filename> file with unpaired reads for single reads library number <#> (<#> =
1,2,...,9)
--trusted-contigs <filename> file with trusted contigs
--untrusted-contigs <filename> file with untrusted contigs](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-196-320.jpg)
![Linha de Comando
Pipeline options:
--only-error-correction runs only read error correction (without assembling)
--only-assembler runs only assembling (without read error correction)
--careful tries to reduce number of mismatches and short indels
--continue continue run from the last available check-point
--restart-from <cp> restart run with updated options and from the specified check-point ('ec', 'as', 'k<int>', 'mc', 'last')
--disable-gzip-output forces error correction not to compress the corrected reads
--disable-rr disables repeat resolution stage of assembling
Advanced options:
--dataset <filename> file with dataset description in YAML format
-t/--threads <int> number of threads
[default: 16]
-m/--memory <int> RAM limit for SPAdes in Gb (terminates if exceeded)
[default: 250]
--tmp-dir <dirname> directory for temporary files
[default: <output_dir>/tmp]
-k <int,int,...> comma-separated list of k-mer sizes (must be odd and
less than 128) [default: 'auto']
--cov-cutoff <float> coverage cutoff value (a positive float number, or 'auto', or 'off') [default: 'off']
--phred-offset <33 or 64> PHRED quality offset in the input reads (33 or 64)
[default: auto-detect]](https://image.slidesharecdn.com/ufmt-montagemdegenomas-191009225354/85/Montagem-de-Genomas-197-320.jpg)








[1] O documento discute técnicas de montagem de genomas a partir de sequências obtidas por sequenciamento de nova geração. [2] É apresentado um overview sobre algoritmos e softwares populares para montagem de genomas, como Newbler, Velvet e SPADES. [3] Também são discutidos conceitos-chave como estratégias de sequenciamento shotgun e hierárquico para reconstrução de sequências genômicas.



































































