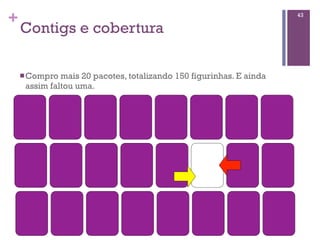
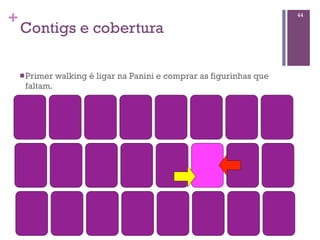
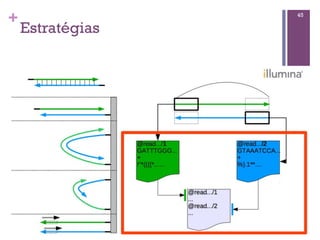
Baixado 52 vezes


































![+
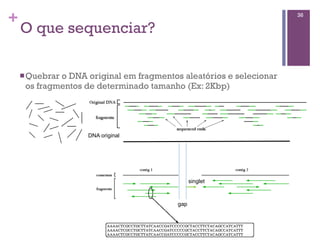
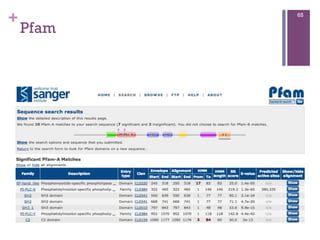
Arquivo de sequência - FASTA
66
>gi|197101743|ref|NP_001125556.1| myoglobin
[Pongo abelii]
MGLSDGEWQLVLNVWGKVEADIPSHGQEVLIRLFKGHPETLEKFDK
FKHLKSEDEMKASEDLKKHGATVLTALGGILKKKGHHEAEIKPLAQ
SHATKHKIPVKYLEFISESIIQVLQSKHPGDFGADAQGAMNKALEL
FRKDMASNYKELGFQG
>gi|386872|gb|AAA59595.1| myoglobin [Homo
sapiens]
MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDK
FKHLKSEDEMKASEDLKKHGATVLTALGGILKKKGHHEAEIKPLAQ
SHATKHKIPVKYLEFISECIIQVLQSKHPGDFGADAEGAMNKALEL
FRKDMASNYKELGFQG](https://image.slidesharecdn.com/minicurso-2013-130911101111-phpapp01/85/Minicurso-2013-67-320.jpg)






























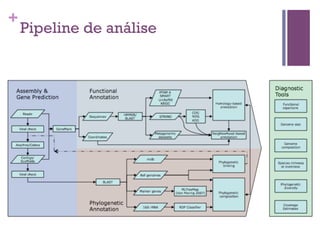
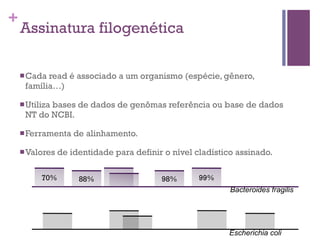
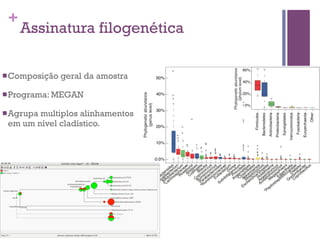
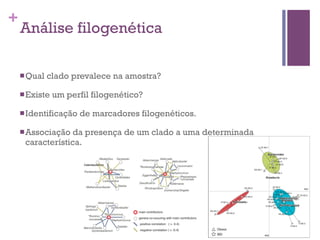
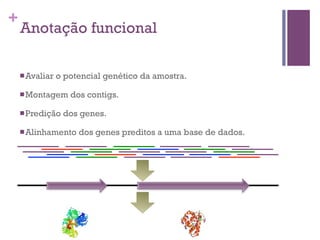


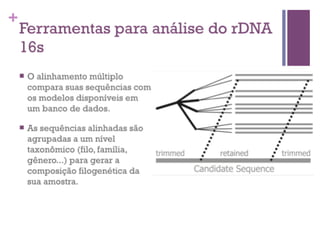
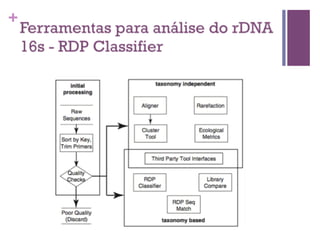


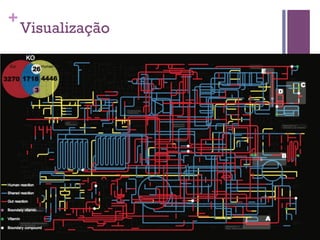

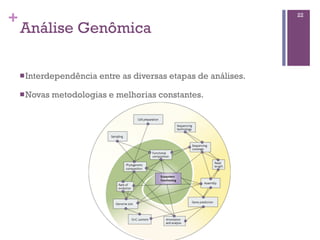
O documento apresenta um resumo sobre bioinformática. Aborda tópicos como a pré-história da bioinformática, a era genômica, ferramentas de análise bioinformática como BLAST e alinhamentos múltiplos, predição de genes e análise funcional.












































![[Especial] 70 edições da Revista Nintendo Blast](https://cdn.slidesharecdn.com/ss_thumbnails/especial70edicoesnintendoblast-150824173659-lva1-app6892-thumbnail.jpg?width=640&height=640&fit=bounds)






















