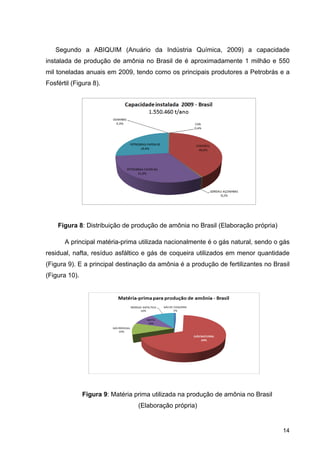
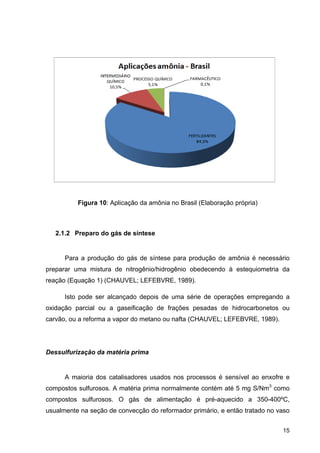
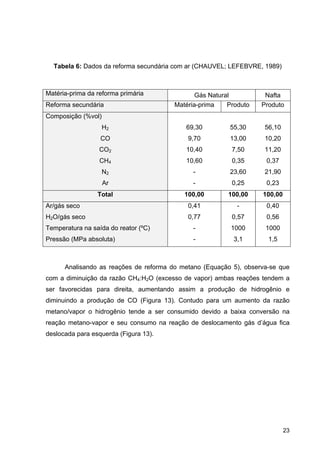
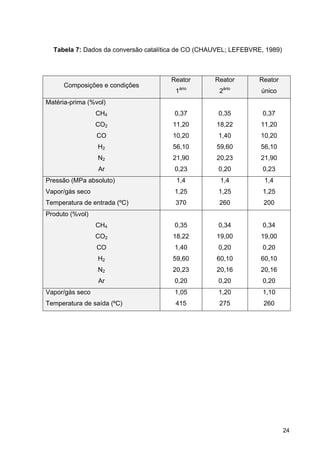
Baixado 93 vezes











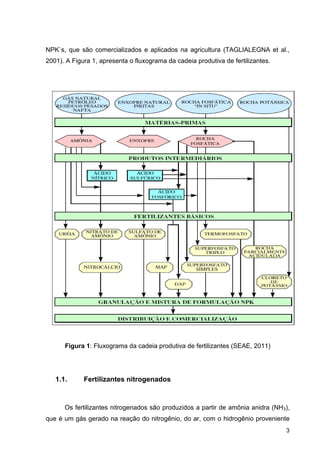
![4
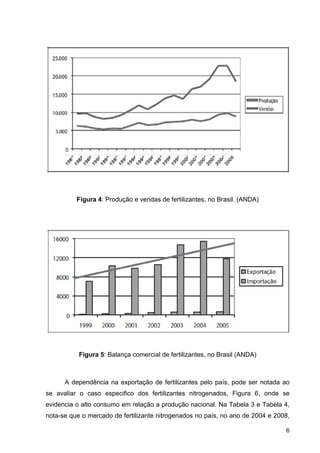
do gás de síntese de diferentes fontes – gás natural, nafta, óleo combustível e outros
derivados de petróleo. Atualmente a fonte de hidrogênio mais utilizada é o gás
natural, devido ao fato deste ser uma melhor fonte de hidrogênio e por gerar um
menor impacto ambiental quando comparado com outras fontes. A Figura 2
apresenta as rotas de produção de alguns fertilizantes nitrogenados.
Figura 2: Produção de fertilizantes nitrogenados (Fertipar)
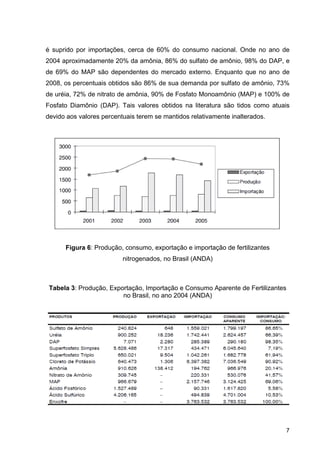
Na Tabela 2 estão presentes os principais fertilizantes nitrogenados
comercializados no Brasil, onde se apresenta o percentual de nitrogênio em função
de diferentes tipos de radicais químicos: nitrato (NO3
-
), amônio (NH4
+
) e uréia
[OC(NH2)2].
Tabela 2: Características químicas dos principais fertilizantes nitrogenados,
comercializados no Brasil. (Fertipar)](https://image.slidesharecdn.com/fertilizantesnitrogenados-140513225215-phpapp01/85/Fertilizantes-nitrogenados-12-320.jpg)

















![31
metanação, como requisitado. A umidade é então removida pela secagem ou
por criogenia.
Metano e argônio não são venenos de catalisador, mas desde que eles são
inertes na reação, eles são susceptíveis de acumular no reciclo de síntese se não
forem removidos por uma purga contínua. Isto pode estar implícito e natural pela
simples dissolução e arraste de amônia produzida, se o conteúdo do gás de
reposição é baixo, menos do que 0,01%, por exemplo, em processos incluindo a
lavagem com nitrogênio liquido. É obrigatório para altas concentrações, que são
acima de 1% se a metanação é realizada, e, neste caso, o gás de reciclo pode
conter mais do que 10% a 15% de metano e argônio. Dependendo das condições de
operação, a vida do catalisador pode ser tão longa quanto dez anos (CHAUVEL;
LEFEBVRE, 1989).
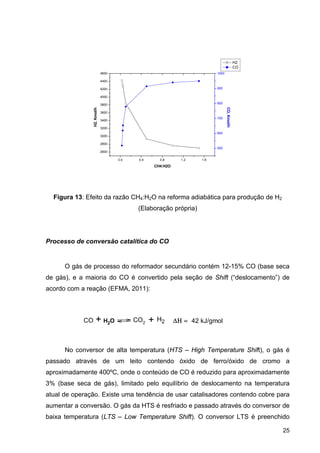
A equação básica mais amplamente aceita para expressar a cinética da
síntese de amônia é aquela de Temkin e Pyzhev (1940). Ela expressa a taxa de
reação como uma função das pressões parciais dos reagentes e produtos:
. ,
,
Equação 10: Cinética da síntese de amônia de Temkin e Pyzhev1
Onde k1 e k2 são as constantes da taxa da síntese e reações de decomposição.
Cálculos precisos devem considerar a atividade dos diferentes componentes
da mistura e a presença do catalisador. Uma das muitas equações derivadas que
considera a influência destes fatores é aquela de Dyson e Simon (1968), que usa a
seguinte expressão (CHAUVEL; LEFEBVRE, 1989):
1
ki=ki0*exp[-Eai/(RT)], onde k10, k20, Ea1, Ea2 ,R são 1,78954.104
kmol.atm-1,5
/(m3
.h),
2,5714.1016
kmol.atm0,5
/(m3
.h), 20800 kcal/kmolN2, 47400 kcal/kmol N2, 1,985887
kcal/kmol.K) respectivamente.](https://image.slidesharecdn.com/fertilizantesnitrogenados-140513225215-phpapp01/85/Fertilizantes-nitrogenados-39-320.jpg)

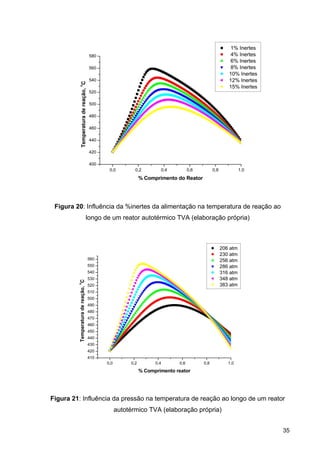



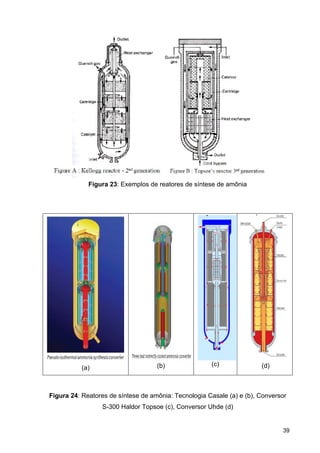
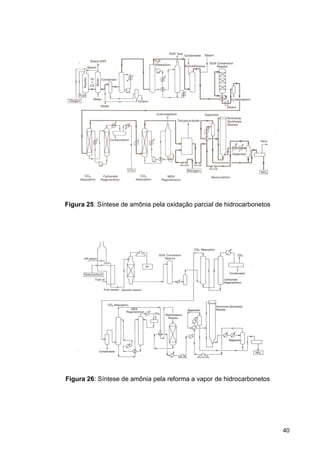
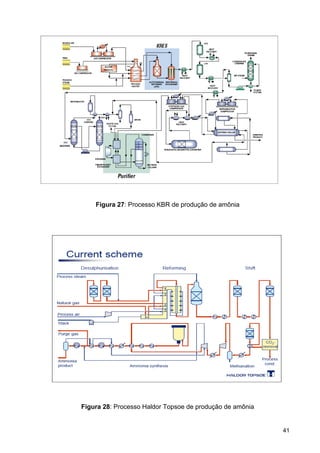
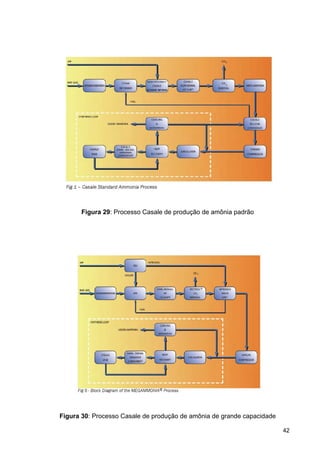
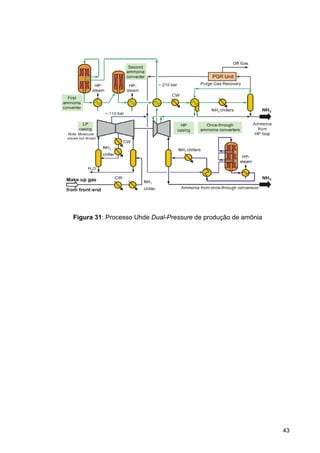







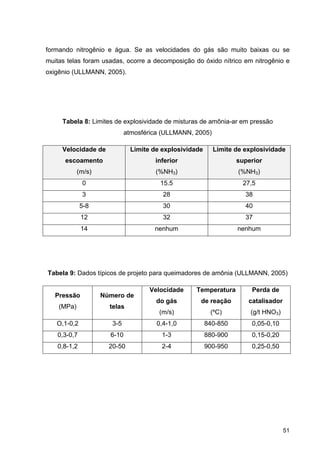
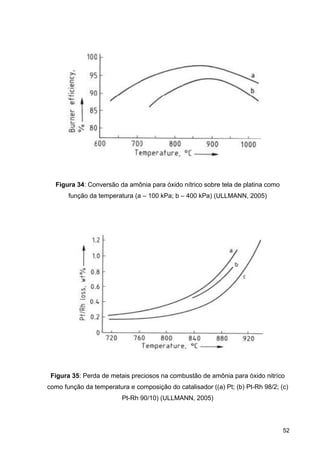
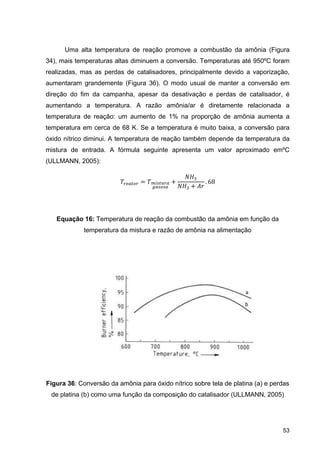








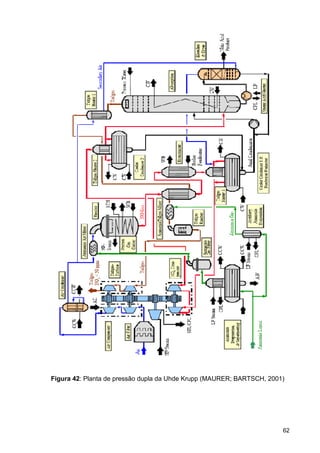
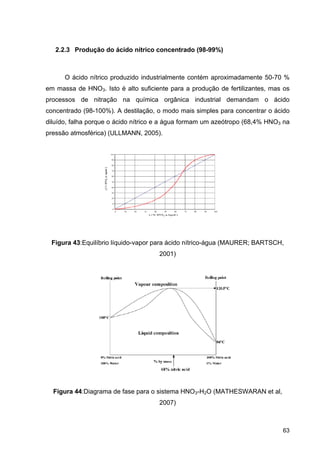
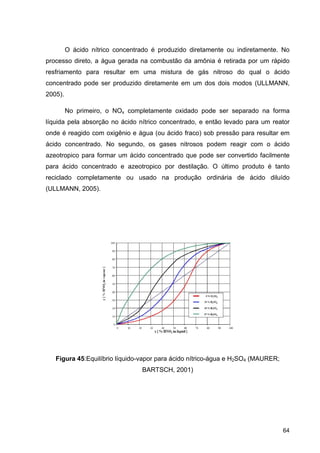












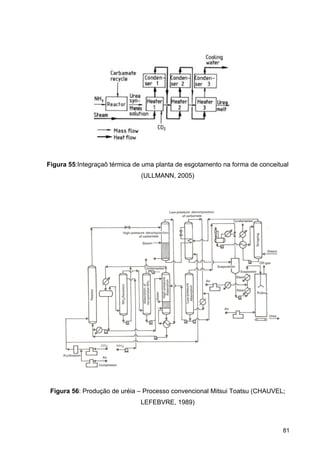
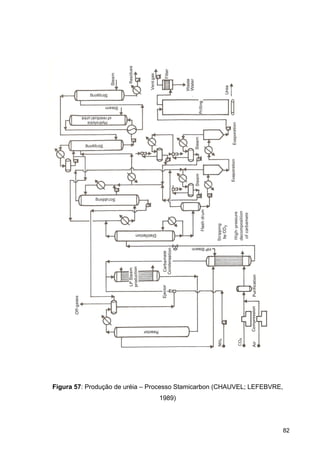






























O documento aborda a produção de fertilizantes nitrogenados, com foco na síntese de amônia e seus derivados, incluindo processos, insumos e a importância econômica desses fertilizantes no Brasil. Apresenta detalhes sobre os processos de produção de ácido nítrico, uréia, nitrato de amônio e sulfato de amônio, além de aspectos termodinâmicos e cinéticos relacionados. A conclusão sintetiza o papel crucial dos fertilizantes nitrogenados na agricultura e na economia nacional.










![[MANO] Introdução a Polímeros - 2a ed.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/manointroduoapolmeros-2aed-220606110423-360a3353-thumbnail.jpg?width=640&height=640&fit=bounds)

















































