Baixado 282 vezes


























































































![90 Farmacopeia Brasileira, 5ª edição
VISCOSÍMETRO DE EFLUXO - MODELO TIPO FORD substâncias opticamente ativas e estas não reagirem entre
si, o ângulo de desvio será a soma algébrica dos ângulos de
Selecionar o orifício adequado. A diretriz para a seleção do desvio de ambas.
orifício deve ser a obtenção de um tempo de escoamento
do líquido em teste ao redor de 60 segundos. Deve-se ter
um tempo de escoamento entre 20 e 100, segundos, para a Polarímetro
amostra a 25 ºC. Historicamente, a polarimetria foi realizada utilizando
A amostra deve ser, perfeitamente, homogeneizada. No um instrumento em que o poder rotatório é estimado pela
momento do ensaio, o viscosímetro e o material a ser visualização da intensidade de campos divididos. Por essa
ensaiado devem estar a 25 ± 0,1 ºC. Fechar o orifício com razão, a linha-D da lâmpada de sódio no comprimento de
lâmina de vidro plana e preencher o copo com amostra onda visível a 589 nm foi o mais frequentemente utilizado.
até o nível mais elevado. Verter a amostra, lentamente, O poder rotatório específico determinado na linha D é
evitando a formação de bolhas. Nivelar a amostra no expresso, na maioria das vezes, pelo símbolo:
copo utilizando placa de vidro plana. Retirar a lâmina do
orifício. A amostra ficará retida dentro do copo. Remova
a placa de vidro plana e acione o cronômetro quando a
amostra começar a escoar pelo orifício. Quando ocorrer
a primeira interrupção do fluxo de escoamento, parar o O uso de comprimentos de onda mais baixos como os
cronômetro e anotar o tempo transcorrido em segundos. disponíveis com as linhas de lâmpada de mercúrio isolados
Realizar o ensaio, no mínimo, em triplicata. A viscosidade por meio de filtros de transmitância máxima a cerca de 578,
será a média dos valores obtidos, expressa em mm2/s ou 546, 436, 405 e 365 nm em um polarímetro fotoelétrico
Centistokes, sendo permitido um desvio padrão máximo mostraram maior sensibilidade; consequentemente houve
3%. A conversão de segundos para mm2/s ou Centistokes uma redução na concentração da substância no ensaio.
5 é dada de acordo com o manual do equipamento utilizado. Em geral, o poder rotatório observado em 436 nm é
aproximadamente o dobro e em 365 nm é cerca de três
vezes maior que o em 589 nm.
5.2.8 DETERMINAÇÃO
DO PODER ROTATÓRIO Poder rotatório e poder rotatório especifico
E DO PODER ROTATÓRIO A equação geral usada em polarimetria é:
ESPECÍFICO
Muitas substâncias farmacêuticas são opticamente ativas, Em que [α] é o poder rotatório específico no comprimento
logo desviam a luz plano-polarizada de modo que a luz de onda λ e na temperatura t, a é o poder rotatório observado
transmitida é desviada em um determinado ângulo em em graus (°), l é o caminho óptico em decímetros e c é a
relação à incidente. concentração da substância em g por 100 mL. Assim, [α]
é 100 vezes α para uma solução contendo 1 g em 100 mL
Substâncias com a mesma estrutura contendo um ou mais
em uma célula com um caminho óptico de 1,0 decímetro
centros quirais as quais são imagens especulares não
sob condições definidas do comprimento de onda da luz
superponíveis uma da outra são denominadas enantiômeros.
incidente e da temperatura.
Um dos enantiômeros desvia a luz plano-polarizada para
a direita (+) e é chamado de dextrógiro, ou d; o antípoda Procedimento
desvia para a esquerda (-) e é conhecido como levógiro
ou l. O ângulo desse desvio é igual em módulo para os O poder rotatório específico de um fármaco é um valor de
enantiômeros, porém com sinais opostos. referência e deve ser calculado a partir do poder rotatório
observado para a solução da amostra ou para o líquido
As propriedades físico-químicas dos enantiômeros como conforme especificado na monografia. As medidas do poder
densidade, índice de refração, momento dipolo-dipolo, rotatório são realizadas a 589 nm a 20 °C exceto quando
pontos de ebulição e fusão são idênticas já que o ambiente especificado. Sempre que um polarímetro fotoelétrico é
químico no qual está inserido cada átomo é igual para os usado, uma única medida corrigida pelo branco é realizada.
enantiômeros. Para um polarímetro visual é empregada a média de pelo
menos cinco determinações corrigidas pelo branco. Em
A polarimetria, isso é, a medição do poder rotatório de
ambos os casos, o solvente utilizado para o preparo da
uma substância com polarímetro é um dos métodos mais
solução da amostra deve ser utilizado como branco ou o tubo
prático para distinguir os enantiômeros e, portanto, é um
vazio no caso de líquidos. A temperatura experimental deve
importante critério de identificação, caracterização e de
ser mantida em ± 0,5 °C em relação ao valor especificado.
determinação de pureza enantiomérica dos fármacos.
Usar o mesmo tubo do polarímetro na mesma orientação
O poder rotatório varia com a temperatura, o comprimento angular para a amostra e o branco. Posicionar o tubo para
de onda da luz incidente, o solvente utilizado, a natureza da que a luz passe por ele na mesma direção cada vez.
substância e sua concentração. Se a solução contiver duas](https://image.slidesharecdn.com/farmacopiabrasileira5ediovolume1-121201102401-phpapp02/85/Farmacopeia-brasileira-5-edicao-volume1-90-320.jpg)




























![Farmacopeia Brasileira, 5ª edição 121
vão de 1 a 10-14 M, que é definida pela simplificada
relação:
pH = - log [H3O+] = log 1/[H3O+],
Desta forma, a escala de pH é uma escala invertida em
relação às concentrações de íon hidrônio, ou seja, quanto
menor a concentração de íon hidrônio, maior o valor do
pH.
A determinação potenciométrica do pH é feita pela
medidada diferença de potencial entre dois eletrodos
adequados, imersos na solução em exame. Um destes
eletrodos é sensível aos íons hidrogênio e o outro é o
eletrodo de referência, de potencial constante.
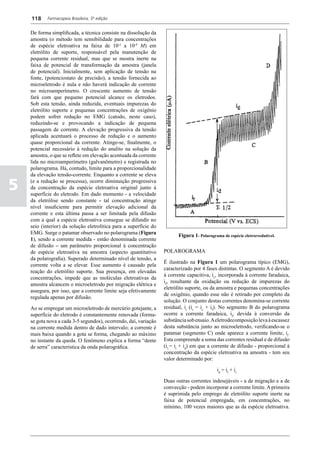
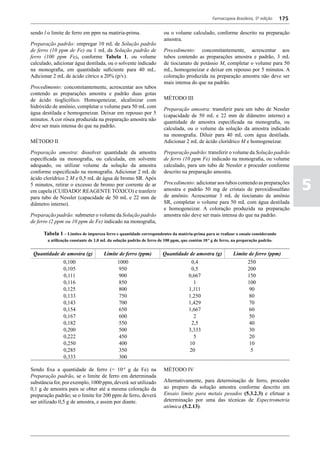
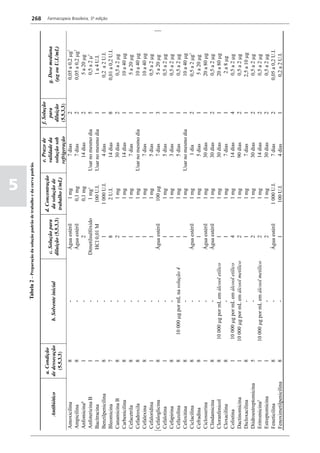
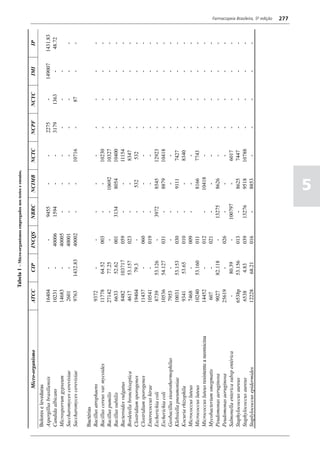
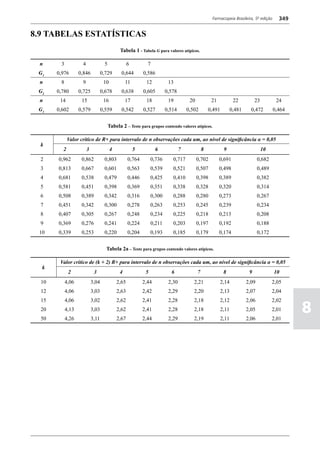
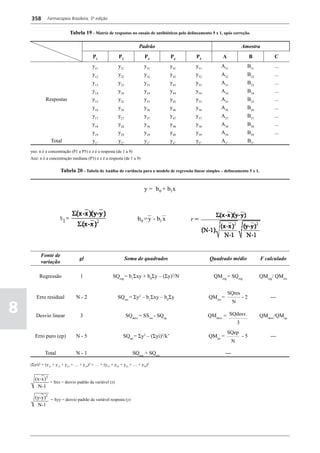
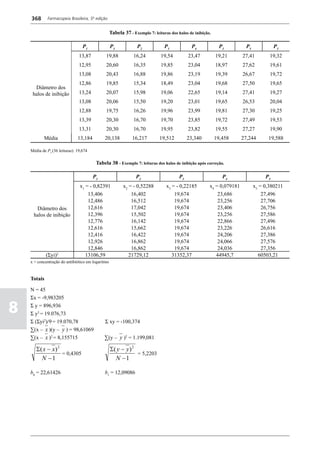
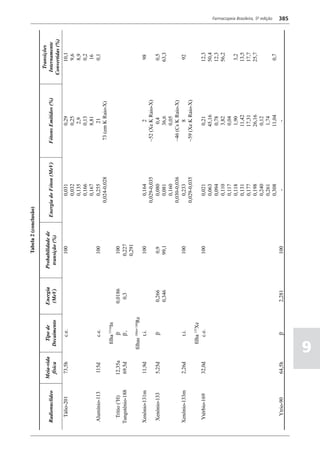
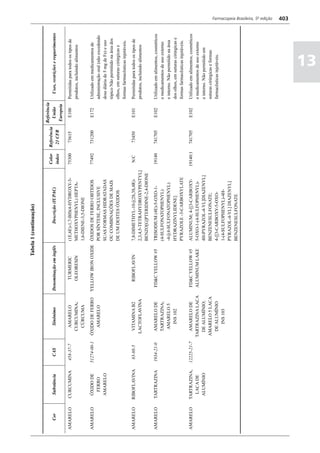
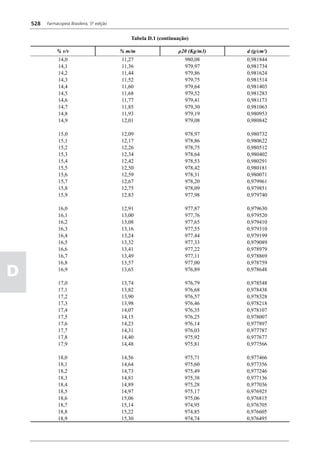
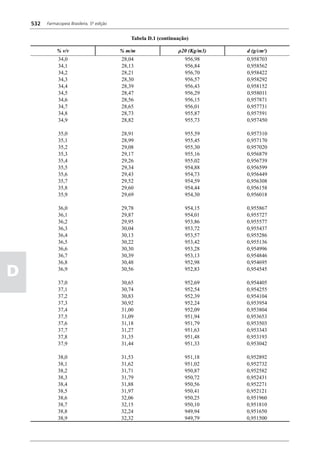
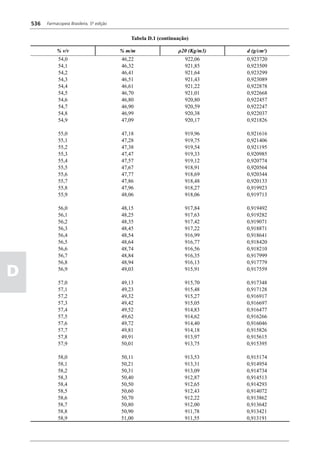
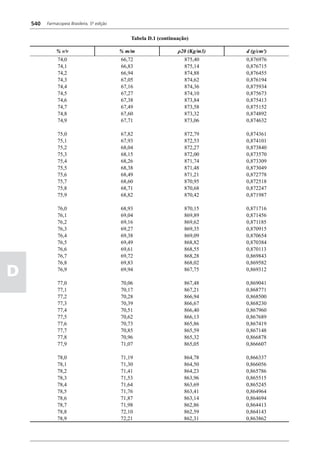
Figura 3 - Polarograma obtido na A equação que expressa a medida potenciométrica de uma
polarografia de pulso diferencial. célula é :
Por outro lado, a técnica de pulsos não provoca diminuição pH = pHt = (E –Et)/ K,
acelerada da camada de difusão (concentração de espécies
eletroativas junto ao eletrodo), propiciando a obtenção de em que
correntes de difusão mais elevadas para concentrações
E = potencial medido quando a célula contém a solução
equivalentes. Daí o aumento de sensibilidade inerente à
amostra,
técnica. Outro aspecto favorável da polarografia de pulso é
a maior facilidade na medida da corrente limite, isenta de
oscilações, ao contrário do que ocorre na polarografia de
Et = potencial medido quando a célula contém a solução
tampão,
pH = valor de pH na solução amostra
5
corrente contínua.
pHt = valor de ph na solução tampão, e
Na polarografia de pulso diferencial, pulsos constantes, K = variação do potencial por unidade de variação de pH -
de pequena amplitude, são sobrepostos a uma rampa de teoricamente equivale a 0,0591631 + 0,000198 (t–25), em
potencial de tensão linearmente crescente. A medida da que t corresponde à temperatura na qual se opera.
corrente é efetuada duas vezes a cada pulso - imediatamente
O valor de pH é expresso pela equação em relação ao pH
antes da aplicação do pulso e, novamente, em seu instante
da solução padrão (pHp) e determinado em peagômetro
final - registrando-se apenas a diferença entre os dois valores
utilizando eletrodo de vidro.
medidos (Figura 2). O registro gráfico deste sistema de
medida diferencial fornece curva semelhante à derivada da Os aparelhos comercialmente utilizados para a
onda polarográfica, mostrando pico característico (Figura determinação de pH são instrumentos potenciométricos,
3). O potencial do pico polarográfico corresponde a E1/2 providos de amplificadores eletrônicos de corrente com
- DE/2 em que DE representa a altura do pico. Graças à célula de vidro-calomelano, os quais são capazes de
natureza do polarograma, que apresenta picos em vez reproduzir valores correspondentes a 0,02 de unidades
de ondas polarográficas tradicionais, a polarografia de de pH. A escala de pH é calibrada não só em milivolts,
pulso diferencial propicia resolução mais elevada, a mas também em unidades correspondentes de pH. Dessa
ponto de permitir determinações simultâneas de espécies forma, não há necessidade de se aplicar a equação acima,
eletroativas com potenciais de meia-onda próximos entre que traduz a medida eletrométrica de pH. Uma vez que as
si, em concentrações da ordem de 10-7 M. medidas de atividade hidrogênica são sensíveis a variações
de temperatura, todos os medidores de pH são equipados
com ajuste eletrônico de temperatura.
5.2.19 DETERMINAÇÃO DO pH
Soluções-tampão para calibração do medidor de pH
DETERMINAÇÃO POTENCIOMÉTRICA DO pH São empregadas visando à aferição do aparelho, permitindo
linearidade nas respostas em relação às alterações de
O valor de pH é definido como a medida da atividade
potencial observadas. As mais importantes são: tetraoxalato
do íon hidrogênio de uma solução. Convencionalmente
de potássio 0,05 M, fosfato equimolar 0,05 M, tetraborato
é usada a escala da concentração de íon hidrogênio da
de sódio 0,01 M, carbonato de sódio e hidróxido de cálcio
solução. A água é um eletrólito extremamente fraco, cuja
saturado a 25 ºC.
autoionização produz íon hidrônio (hidrogênio hidratado)
e íon hidróxido: As soluções-tampão são preparadas da seguinte maneira:
H2O + H2O H3O+ + OH- Tetraoxalato de potássio, 0,05 M – Reduzir o tetraoxalato
de potássio a pó fino e dessecar em dessecador com sílica.
As concentrações do íon hidrônio nas soluções aquosas
Dissolver exatamente 12,71g de KH3(C2O4)∙2H2O em água
podem variar entre limites amplos, que experimentalmente
para perfazer 1000 ml.](https://image.slidesharecdn.com/farmacopiabrasileira5ediovolume1-121201102401-phpapp02/85/Farmacopeia-brasileira-5-edicao-volume1-121-320.jpg)



































![Farmacopeia Brasileira, 5ª edição 159
5.2.29.16 DETERMINAÇÃO DE ESTERÓIS colesterol e à betulina. Os cromatogramas obtidos com
EM ÓLEOS FIXOS as Soluções amostra apresentam bandas de Rf próximos
dos correspondentes aos esteróis. De cada um dos
cromatogramas raspar a região da placa correspondente às
bandas dos esteróis bem como uma zona situada 2 - 3 mm
SEPARAÇÃO DA FRAÇÃO DE ESTERÓIS
para cima e para baixo das zonas visíveis correspondentes
Preparar a fração insaponificável. Separar a fração de à solução padrão. Colocar essas regiões em três erlenmeyer
esteróis do óleo fixo por cromatografia em camada diferentes de 50 mL. Adicionar a cada um, 15 mL de
delgada, utilizando uma placa de gel de sílica G (espessura diclorometano quente e agitar. Filtrar, separadamente, cada
da camada entre 0,3 mm e 0,5 mm). solução por um filtro de vidro poroso (40), ou por filtro
de papel apropriado. Lavar, cada filtro, três vezes com 15
Solução amostra (a). Em balão de 150 mL, introduzir mL de diclorometano. Transferir o filtrado e líquidos de
volume de solução de betulina a 2 g/L em diclorometano, que lavagem em erlenmeyer, tarado. Evaporar até a secura em
corresponda a cerca de 10% do teor de esteróis da amostra corrente de nitrogênio e pesar.
utilizada para o doseamento (por exemplo, volume de 500 μL
de solução de betulina no caso do óleo de oliva virgem, e de
DOSEAMENTO DOS ESTERÓIS
1500 μL no caso de outros óleos vegetais). Se na monografia
estiver registrada a exigência do cálculo do teor percentual Proceder por cromatografia a gás (5.2.17.5). O doseamento
de cada esterol na fração esterólica, a adição da betulina pode deve ser realizado ao abrigo da umidade e preparar as
ser omitida. Evaporar até a secura em corrente de nitrogênio. soluções no momento do uso.
Adicionar 5,00 g da amostra e adicionar 50 mL de hidróxido
de potássio alcoólico 2 M. Acoplar condensador de refluxo Solução amostra. Aos esteróis separados a partir da amostra
vertical. Aquecer em banho-maria durante 1 h, sob agitação. por cromatografia em camada delgada, adicionar 0,02 mL
5
Arrefecer até temperatura inferior a 25 °C e transferir o da mistura, recentemente preparada, de clorotrimetilsilano:
conteúdo do balão para um funil de separação, com o auxílio hexametildissilazano : piridina anidra (1:3:9; v/v/v)
de 100 mL de água. Agitar, com precaução, três vezes por miligrama de resíduo. Agitar, cuidadosamente, até
com 100 mL de éter etílico isento de peróxidos. Reunir as dissolução completa dos esteróis. Deixar em repouso em
frações etéreas em outro funil de separação com o auxílio dessecador com pentóxido de difósforo durante 30 min.
de 40 mL de água destilada. Agitar suavemente durante Centrifugar, se necessário, e utilizar o sobrenadante.
alguns minutos. Deixar separar as fases por decantação e
rejeitar a fase aquosa. Lavar a fase orgânica várias vezes Solução padrão (a). A nove partes dos esteróis separados
com 40 mL de água (a cada vez) até que a fase aquosa não do óleo de canola por cromatografia em camada delgada,
apresente reação alcalina a fenolftaleína. Transferir a fração juntar uma parte de colesterol. Adicionar 0,02 mL da
orgânica para um balão, tarado e lavar o funil de separação mistura, recentemente preparada, de clorotrimetilsila
com éter etílico. Evaporar o éter. Adicionar ao resíduo 6 no:hexametildissilazano:piridina anidra (1:3:9; v/v/v)
mL de acetona. Eliminar, cuidadosamente, o solvente com por miligrama de resíduo. Agitar, cuidadosamente, até
corrente de nitrogênio. Secar em estufa a 100 - 105 °C até dissolução completa dos esteróis. Deixar em repouso em
massa constante. Dissolver o resíduo com volume mínimo dessecador com pentóxido de difósforo durante 30 min.
de diclorometano. Centrifugar, se necessário, e utilizar o sobrenadante.
Solução amostra (b).Submeter 5,00 g de óleo de canola Solução padrão (b). Aos esteróis separados do óleo
ao mesmo procedimento descrito para a Solução amostra de girassol por cromatografia em camada delgada,
(a) a partir de “Adicionar 50 mL de hidróxido de potássio juntar 0,02 mL da mistura, recentemente preparada,
alcoólico 2 M...”. de clorotrimetilsilano : hexametildissilazano : piridina
anidra (1:3:9; v/v/v) por miligrama de resíduo. Agitar,
Solução problema (c). Submeter 5,00 g de óleo de girassol cuidadosamente, até dissolução completa dos esteróis.
ao mesmo procedimento descrito para a Solução problema Deixar em repouso em dessecador com pentóxido de
(a) a partir de “Adicionar 50 mL de hidróxido de potássio difósforo durante 30 min. Centrifugar, se necessário, e
alcoólico 2 M...”. utilizar o sobrenadante.
Solução padrão. Dissolver 25 mg de colesterol e 10 mg
de betulina em 1 mL de diclorometano. Utilizar uma placa Condições cromatográficas
diferente para cada solução problema. - coluna de sílica fundida de 20 a 30 m de comprimento e
Aplicar, separadamente, 20 μL da Solução padrão em 0,25-0,32 mm de diâmetro interno, recoberta por película
forma de banda de 20 mm por 3 mm e 0,4 mL da solução de poli[metil(95)fenil(5)]siloxano ou de poli[metil(94)
problema (a), (b) ou (c) em forma de banda de 40 mm por fenil(5)vinil(l)]siloxano (espessura da película 0,25 μm);
3 mm. Migrar pela distância de 18 cm com a fase móvel - gás de arraste: gás hidrogênio com fluxo de 30 a 50 cm/s
éter:n-hexano (35:65; v/v). Secar as placas em corrente de ou hélio com fluxo de 20 a 35 cm/s;
nitrogênio. Revelar com solução de diclorofluoresceína a 2 - razão de split (1/50 ou 1/100);
g/L em etanol. Examinar em 254 nm. - temperaturas: coluna: 260 °C; injetor: 280 °C; detector:
290 °C;
O cromatograma obtido com a Solução padrão - volume de injeção: 1 μL.
apresenta bandas correspondentes, respectivamente, ao](https://image.slidesharecdn.com/farmacopiabrasileira5ediovolume1-121201102401-phpapp02/85/Farmacopeia-brasileira-5-edicao-volume1-159-320.jpg)




































































































































































































































































































































1. Este documento é a 5a edição da Farmacopeia Brasileira, publicada em 2010 pela Agência Nacional de Vigilância Sanitária (Anvisa). 2. A Farmacopeia Brasileira estabelece normas e especificações para insumos farmacêuticos, medicamentos e outros produtos sujeitos à vigilância sanitária no Brasil. 3. Esta edição revisa e atualiza as monografias e métodos gerais das edições anteriores e inclui novas informações, totalizando 176 métodos gerais e 599 monograf



































![Guia de qualidade_para_sistemas[1]](https://cdn.slidesharecdn.com/ss_thumbnails/guiadequalidadeparasistemas1-150612184139-lva1-app6891-thumbnail.jpg?width=640&height=640&fit=bounds)



















































