Baixado 64 vezes




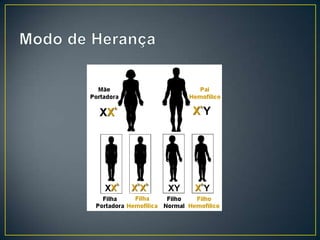



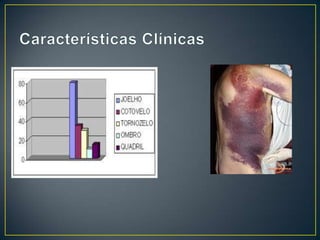







O documento resume as principais características da hemofilia A, um distúrbio hereditário da coagulação causado pela deficiência do fator VIII. A hemofilia A causa sangramentos espontâneos, principalmente em articulações, e é mais comum em homens. O tratamento envolve a reposição do fator VIII através de derivados plasmáticos ou recombinantes.





![[c7s] Hemofilia](https://cdn.slidesharecdn.com/ss_thumbnails/hemofilia-111129143931-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)






































![Hemofilia[2]](https://cdn.slidesharecdn.com/ss_thumbnails/hemofilia2-1198935942535703-5-thumbnail.jpg?width=640&height=640&fit=bounds)






















