Baixado 27 vezes



















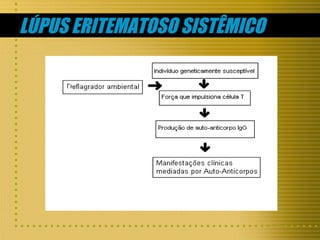



















O documento descreve as principais células do sistema imunológico, incluindo linfócitos T e B, macrófagos e células NK. Também discute doenças da imunidade como hipersensibilidade, doenças autoimunes como lúpus e síndrome de Sjögren, e rejeição de transplantes.



































































