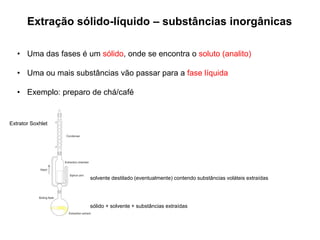
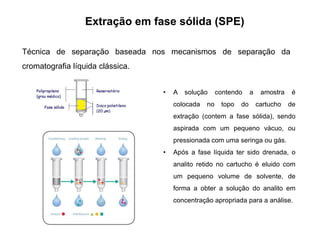
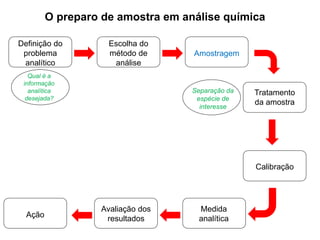
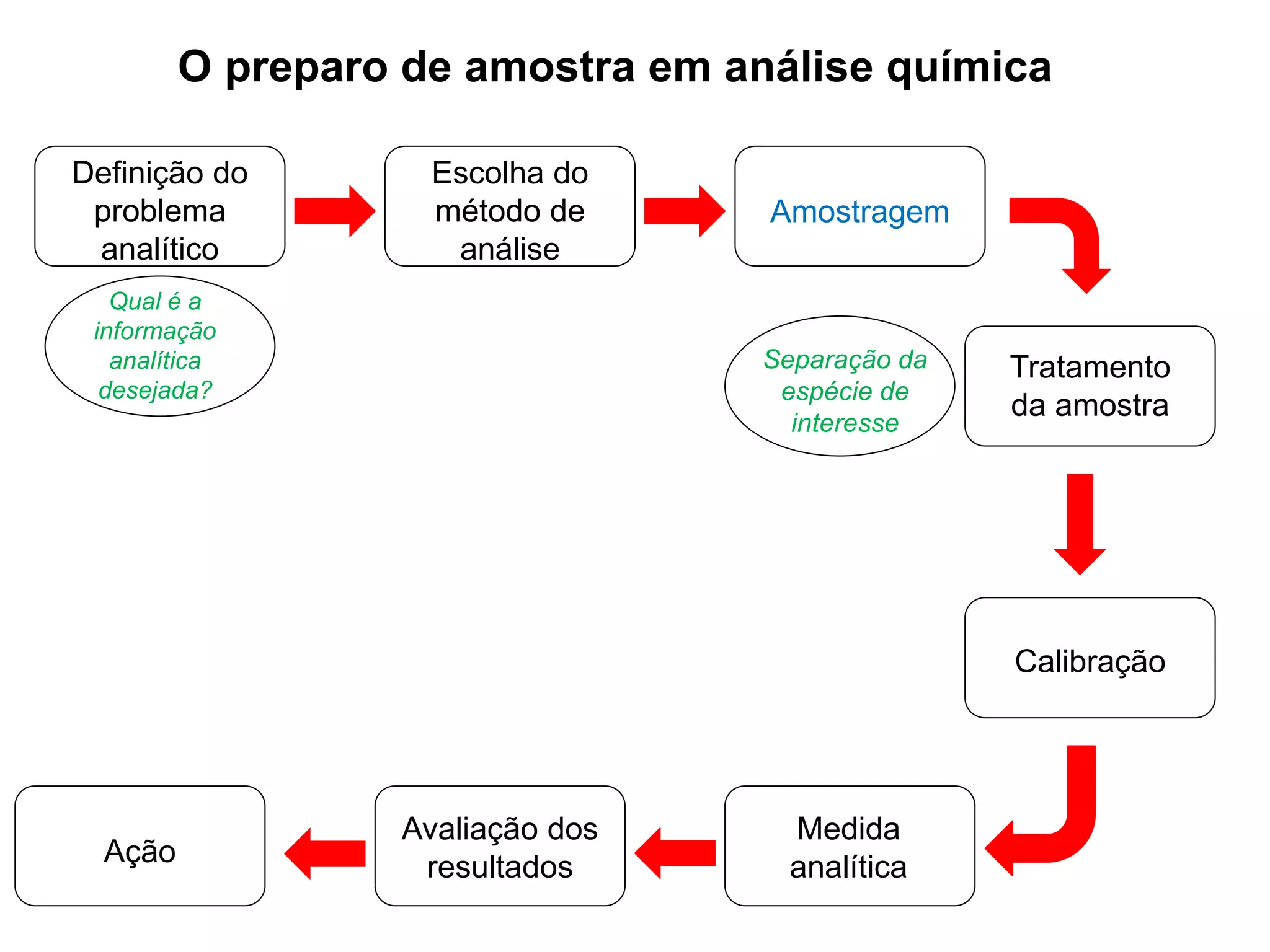
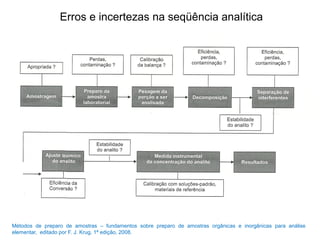
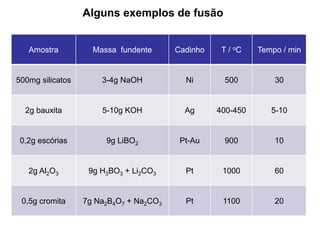
[1] O documento discute o processo de preparo de amostras para análise química, incluindo etapas como amostragem, tratamento da amostra, separação da espécie de interesse e calibração.
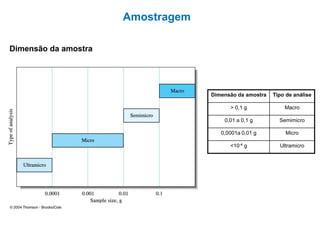
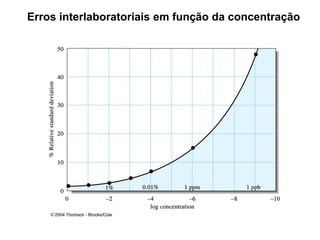
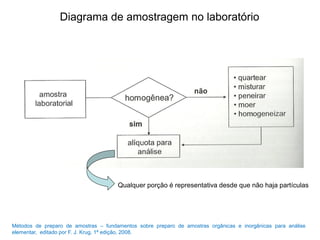
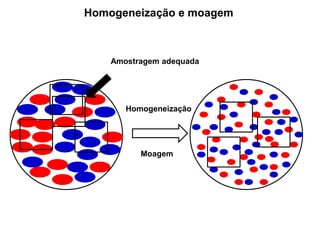
[2] A amostragem é o processo de selecionar e remover uma pequena porção representativa do material a ser analisado. Erros na amostragem podem comprometer a precisão dos resultados.
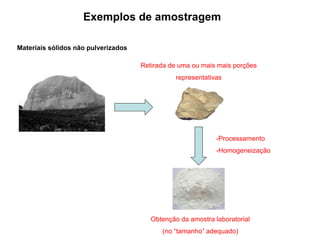
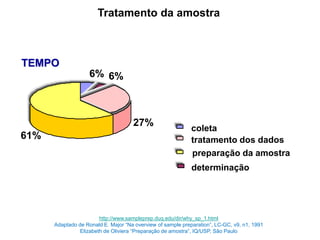
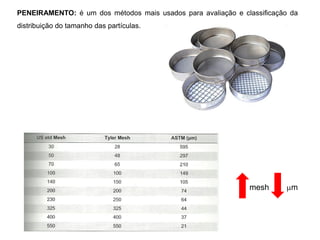
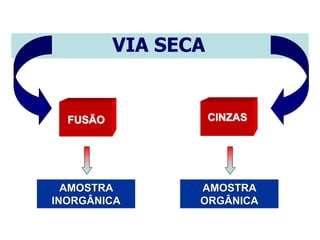
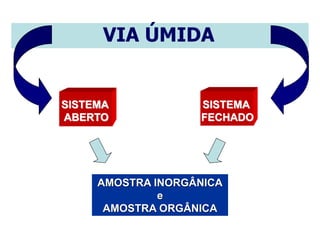
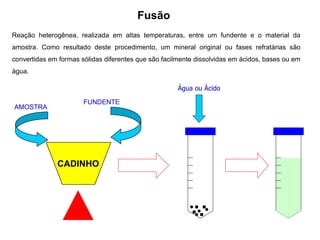
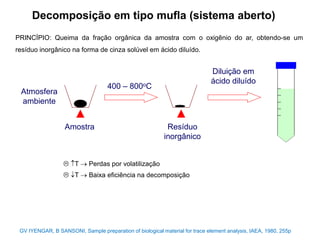
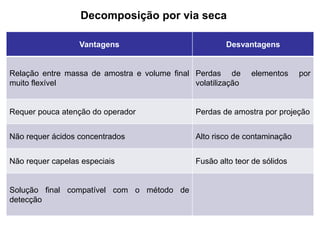
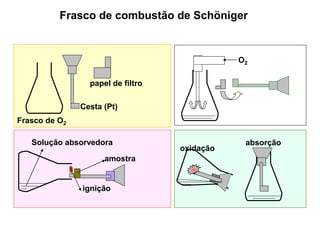
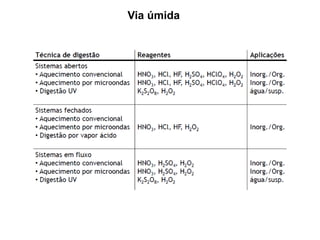
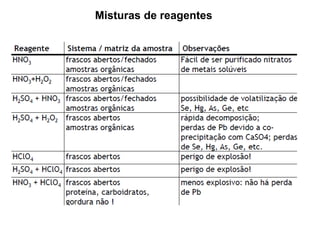
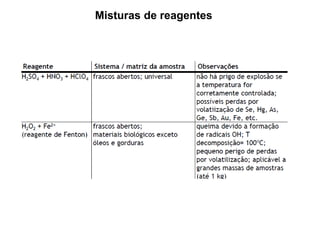
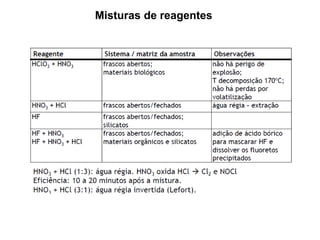
[3] O tratamento da amostra geralmente envolve processos como secagem, moagem e dissolução para converter










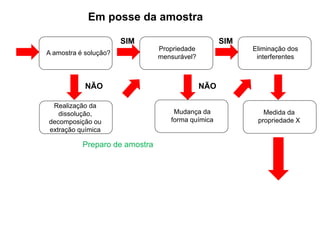







































![Princípios
A partição de um soluto entre 2 fases líquidas imiscíveis é um fenômeno de
equilíbrio governado pela LEI DE DISTRIBUIÇÃO.
Se o soluto da espécie A distribui-se entre a água e uma fase orgânica, o
equilíbrio resultante pode ser descrito como:
A(aq) ⇌ A(org)
A razão para as atividades para A nas 2 fases será uma constante e
independente da quantidade total de A, a uma dada T:
aq
org
aq
A
org
A
A
A
a
a
K
]
[
]
[
)
(
)
(
Coeficiente de
partição ou
distribuição
Extração de
uma solução
aquosa com
solvente
orgânico](https://image.slidesharecdn.com/aula-10-preparo-de-amostras-230417201644-eeb2b49f/85/aula-10-Preparo-de-amostras-pdf-77-320.jpg)