1. A medicina é uma ciência em constante evolução, exigindo modificações no tratamento e farmacoterapia à medida que novas pesquisas e experiência clínica ampliam o conhecimento.
2. Os organizadores consultaram fontes confiáveis para oferecer informações completas e de acordo com os padrões aceitos na época da publicação.
3. Entretanto, devido à possibilidade de erros humanos ou alterações nas ciências médicas, os leitores devem confirmar as informações com outras fontes e conferir a b


![Nota
'
A medicina é uma ciência em constante evolução. A medida que novas pesquisas e a experiência clínica ampliam
o nosso conhecimento, são necessárias modificações no tratamento e na farmacoterapia. Os organizadores desta
obra consultaram as fontes consideradas confiáveis, em um esforço para oferecer informações completas e, geral
mente, de acordo com os padrões aceitos à época da publicação. Entretanto, tendo em vista a possibilidade de falha
humana ou de alterações nas ciências médicas, os leitores devem confirmar estas informações com outras fontes.
Por exemplo, e em particular, os leitores são aconselhados a conferir a bula de qualquer medicamento que preten
dam administrar, para se certificarde que a informação contida neste livro está correta e de que não houve alteração
na dose recomendada nem nas contraindicações para o seu uso. Esta recomendação é particularmente importante
em relação a medicamentos novos ou raramente usados.
F233 Farmacologia ilustrada [recurso eletrônico] / Michelle A.
Clark ... [et ai.] ; tradução e revisão técnica: Augusto
Langeloh. - 5.ed.- Dados eletrônicos. - Porto Alegre :
Artmed, 2013.
Editado tabém como livro impresso em 2013.
ISBN 978-85-65852-69-2
1 . Farmacologia ilustrada. 1. Clark, Michelle A.
CDU 615-028.22
Catalogação na publicação: Ana Paula M. Magnus - CRB 10/2052](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-2-320.jpg)








![1. RESUMO
. , .
acoc 1 ne 1ca
A farmacocinética estuda o que o organismo faz com o fármaco, ao passo
que a farmacodinâmica (ver Capítulo 2) descreve o que o fármaco faz no or
ganismo. Uma vez administrado por uma das várias vias disponíveis, quatro
propriedades farmacocinéticas determinam a velocidade do início da ação, a
intensidade do efeito e a duração da ação do fármaco (Figura 1 .1):
• Absorção: primeiro, a absorção do fármaco desde o local de adminis
tração (absorção) permite o acesso do agente terapêutico (seja direta
ou indiretamente) no plasma.
• Distribuição: segundo, o fármaco pode, então, reversivelmente, sair da
circulação sanguínea edistribuir-se nos líquidos intersticial e intracelular.
• Biotransformação ou metabolismo: terceiro, o fármaco pode ser bio
transformado no fígado ou em outros tecidos.
• Eliminação: finalmente, o fármaco e seus metabólitos são eliminados
do organismo na urina, na bile ou nas fezes.
As variáveis farmacocinéticas permitem ao clínico elaborar e otimizar os regi
mes terapêuticos, incluindo as decisões quanto àvia de administração de cada
fármaco, à quantidade e à frequência de cada dose e a duração do tratamento.
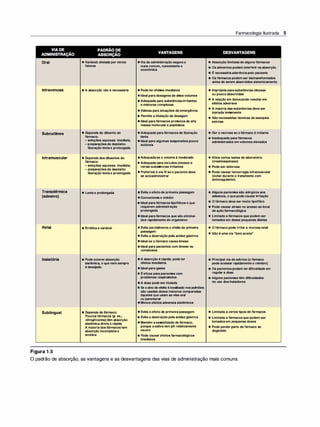
li. VIAS DE ADMINISTRAÇÃO DE FÁRMACOS
A via de administração é determinada primariamente pelas propriedades do
fármaco (p. ex., hidra ou lipossolubilidade, ionização) e pelos objetivos tera
pêuticos (p. ex., a necessidade de um início rápido de ação, a necessidade de
tratamento por longo tempo, ou a restrição de acesso a um local específico).
As vias principais de administração de fármacos incluem a enteral, a parente
ral e a tópica, entre outras. A Figura 1 .2 ilustra as subcategorias dessas vias,
bem como outros métodos de administração de fármacos.
( Fármaco no local da administração
)
Absorção
(entrada)
(Fármaco no plasma
)
�aDistribuição ]
Fármaco nos tecidos
iEIBiotransformaçãoJ
Metabólito(s) nos tecidos
Excreção
(saída)
Fármaco e metabólito(s)
na urina, na bile ou nas fezes
Figura 1 .1
Representação esquemática da absor
ção, distribuição, biotransformação e
-
excreçao.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-11-320.jpg)




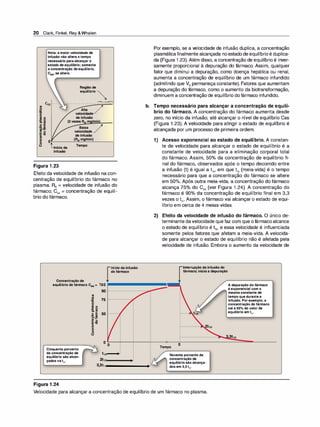
![membrana celular. Na endocitose, as moléculas do fármaco são
engolfadas pela membrana e transportadas para o interior da cé
lula pela compressão da vesícula cheia de fármaco (Figura 1 .6D).
A exocitose é o inverso da endocitose e é usada pelas células para
secretarvárias substâncias por um processo similarao da formação
de vesículas. A vitamina 812 é transportada através da parede in
testinal por endocitose enquanto certos neurotransmissores (p. ex.,
norepinefrina) são armazenados em vesículas intracelulares ligadas
à membrana no terminal nervoso e liberados por exocitose.
B. Fatores que influenciam a absorção
1. Efeito do pH na absorção de fármacos. A maioria dos fármacos é
ácido fraco ou base fraca. Fármacos ácidos (HA) liberam um próton
(H+) causando a formação de um ânion (A-)
2
:
As bases fracas (8H+) também podem liberar um H+. Contudo, a
forma protonada dos fármacos básicos, em geral, é carregada, e a
perda do próton produz a base (8) não ionizada:
Um fármaco atravessa a membrana mais facilmente se ele estiver não
ionizado (Figura 1.7).Assim, para os ácidos fracos, aforma HA não io
nizada consegue permearatravés das membranas, mas oA- nãocon
segue. Para a base fraca, a forma não ionizada, 8, consegue penetrar
através das membranas celulares, contudo o 8H+ protonado não con
segue. Por isso, a concentração efetiva da forma permeável de cada
fármaco no seu local de absorção é determinada pelas concentrações
relativas entre as formas ionizada e não ionizada. A relação entre as
duasformasé, porsuavez, determinada pelo pH no local de absorção
e pela força do ácido ou base fracos, que é representada pela cons
tante de ionização, o pKª (Figura 1.8). (Nota: o pKª é uma medida da
força da interação de um composto com um próton. Quanto menor o
pKª de um fármaco, mais ácido ele é; quanto maior o pK8, mais bá
sico é o fármaco.) O equilíbrio de distribuição é alcançado quando a
2
Ver Bioquímica /lustrada, 4ª edição, Artmed Editora, para uma discussão
sobre equilíbrio ácido-base.
Farmacologia Ilustrada 7
mÁcido fraco
Membrana
lipídica
.
..
..
..
•
•••
+ A- ·.·.
H ..... . 1
� ··.::::
H
<>
o
o
o
o
o
o
o
g
<>
<>
i
Compartimento �
corporal & -
ª
-
Compartimento
corporal
l]Jease fraca
Membrana
o
<>
o
<>
g
<>
g
<>
g
<>
§
Compartimentol
corporal &
Figura 1.7
lipídica
Compartimento
corporal
A. Difusão da forma não ionizadade um
ácido fraco através da membrana lipídi
ca. B. Difusão da forma não ionizada de
uma base fraca através da membrana
lipídica.
QuandoopHémenordoqueo
pKa,asformasprotonadas
HAeBH+predominam QuandopH= pKa,
[HA]=[A-]e
[BH+]= [B]
QuandoopHémaiordoqueo
pKa,asformasdesprotonadas
A-eBpredominam
�-....... ---�
2 pH 3 4 5 6 7 8 9 10 11
PKa
Figura 1.8
A distribuição de um fármaco entre sua forma ionizada e não ionizada depende do pH do ambiente e do pKª do
fármaco. Para exemplificar, o fármaco nesta figura foi imaginado com um pKª de 6,5.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-17-320.jpg)
![8 Clark,Finkel,Rey& Whalen
1 1 11 1
ATP
Figura 1.9
Fármaco{extracelular)
1 11 '
Fármaco{intracelular)
As seis alças transmembrana da gli
coproteína-P formam um canal central
para o bombeamento de fármacos de
pendente de ATP para fora da célula.
forma permeável de um fármaco alcança uma concentração igual em
todos os espaços aquosos do organismo. (Nota: fármacos altamente
lipossolúveis atravessam de modo rápido a membrana celular e, em
geral, entram nos tecidos com uma velocidade determinada pelofluxo
sanguíneo.)
2. Fluxo de sangue no local de absorção. Como o fluxo de sangue
para o intestino é muito maior do que ofluxo para o estômago, a ab
sorção no intestino é maior do que a que ocorre no estômago. (Nota:
o choque reduz drasticamente o fluxo sanguíneo aos tecidos cutâ
neos, minimizando a absorção de administrações subcutâneas.)
3. Área ou superfície disponível para absorção. Com uma superfície
rica em bordas de escova contendo microvilosidades, o intestino tem
uma superfície cerca de 1 .000 vezes maior do que a do estômago,
tornando a absorção de fármacos pelo intestino mais eficiente.
4. Tempo de contato com a superfície de absorção. Se um fármaco
se desloca muito rápido ao longo do TGI, como pode ocorrer em
uma diarreia intensa, ele não é bem absorvido. Contudo, qualquer
retardo no transporte do fármaco do estômago para o intestino re
duz a sua velocidade de absorção. (Nota: o tônus parassimpático
acelera o esvaziamento gástrico, e o tônus simpático [p. ex., cau
sado pelo exercício ou por emoções estressantes] e os anticolinér
gicos [p. ex., diciclomina] o retarda. Assim, a presença de alimento
no estômago dilui o fármaco e retarda o esvaziamento gástrico. Por
tanto, quando um fármaco é ingerido com o alimento, em geral, será
absorvido mais lentamente.)
5. Expressão da glicoproteína-P. A glicoproteína P é uma proteína
transportadora transmembrana para vários fármacos, sendo res
ponsável pelo transporte de várias moléculas, incluindo fármacos,
através da membrana celular (Figura 1 .9). É expressa por todo o
organismo e suas funções incluem:
• Nofígado:transportarfármacos paraa bilevisandoàsuaeliminação
• Nos rins: bombear fármacos para a urina visando à excreção
• Na placenta: transportar fármacos de volta para o sangue mater
no, reduzindo, assim, a exposição do feto aos fármacos
• No intestino: transportar fármacos para o lúmen intestinal e redu
zir a absorção
• Nos capilares do cérebro: bombear os fármacos de volta ao san-
gue, limitando seu acesso ao cérebro
Assim, nas áreas de expressão elevada, a glicoproteína-P diminui
a absorção de fármacos. Além de transportar vários fármacos para
fora das células, ela também está associada com a resistência a
vários fármacos (ver p. 485).
C. Biodisponibilidade
Biodisponibilidade é a fração do fármaco administrado que alcança a
circulação sistêmica. Por exemplo, se 100 mg de um fármaco forem ad
ministrados por via oral e 70 mg desse fármaco forem absorvidos inal
terados, a biodisponibilidade será 0,7 ou 70°
/
o. Determinar a biodisponi
bilidade é importante para calcular a dosagem de fármaco para vias de
administração não IV. A via de administração do fármaco, bem como as
suas propriedades físicas e químicas afetam sua biodisponibilidade.
1. Determinação de biodisponibilidade. A biodisponibilidade é de
terminada pela comparação dos níveis plasmáticos do fármaco de-](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-18-320.jpg)

![1O Clark, Finkel, Rey &Whalen
1 ,5
ri 1,25
·
-
...
'GI
UI
1
o
1111
�
J:I 0,75
fi
� 0,5 ._._,__
°
Fase de
º·25 distribuição
o 1
'+-ln'
Figura 1.12
Fase de
eliminação
2 3
Tempo
4
Concentrações dofármaco no soro após
uma injeção única do fármaco. Admite
se que o fármaco se distribui e subse
quentemente é eliminado.
E. Equivalência terapêutica
Dois medicamentos são terapeuticamente iguais se eles são farmaceu
ticamente equivalentes com perfis clínicos e de segurança similares.
(Nota: a eficácia clínica com frequência depende da concentração sérica
máxima e do tempo necessário [após a administração] para alcançar o
pico de concentração. Portanto, dois fármacos que são bioequivalentes
podem não ser equivalentes terapeuticamente.)
IV. DISTRIBUIÇÃO DE FÁRMACOS
Distribuição de fármacos é o processo pelo qual um fármaco reversivelmente
abandona o leito vascular e entra no interstício (líquido extracelular) e, então,
nas células dos tecidos. Para o fármaco administrado por via IV, quando não
existe a absorção, a fase inicial (isto é, desde imediatamente após a adminis
tração até a rápida queda na concentração) representa a fase de distribuição,
na qual o fármaco rapidamente desaparece da circulação e entra nos tecidos
(Figura 1 .12). Isto é seguido da fase de eliminação (ver p. 13), quando o fár
maco no plasma está em equilíbrio com o fármaco nos tecidos. A passagem
do fármaco do plasma ao interstício depende principalmente do débito car
díaco e do fluxo sanguíneo regional, da permeabilidade capilar, do volume do
tecido, do grau de ligação do fármaco às proteínas plasmáticas e tissulares e
da hidrofobicidade relativa do fármaco.
A. Fluxo sanguíneo
A velocidade do fluxo sanguíneo nos tecidoscapilaresvaria bastante como
resultado de uma distribuição desigual do débito cardíaco aos vários ór
gãos. O fluxo sanguíneo para o cérebro, para o fígado e para os rins é
maior do que para os músculos esqueléticos. O tecido adiposo, a pele e
as vísceras têm fluxo sanguíneo ainda menor. A variação no fluxo de san
gue explica parcialmente a curta duração da hipnose produzida por um
bolus de injeção IV de propofol (ver p. 144). O elevado fluxo sanguíneo,
junto com a elevada lipossolubilidade do propofo/, permite-lhe mover-se
de modo rápido para o SNC e produzir anestesia. A subsequente e lenta
distribuição aos músculos esqueléticos e ao tecido adiposo diminui a con
centração plasmática de modo suficiente, de forma que a concentração
elevada no SNC se reduz e assim se recupera a consciência.
B. Permeabilidade capilar
A permeabilidade capilar é determinada pela estrutura capilar e pela na
tureza química do fármaco. A estrutura capilar varia bastante em termos
de fração da membrana basal, que é exposta pelas junções com frestas
entre as células endoteliais. No fígado e no baço, grande parte da mem
brana basal é exposta a capilares descontínuos e grandes, através dos
quais podem passar grandes proteínas plasmáticas (Figura 1 .13A). Isso
contrasta com o cérebro, onde a estrutura capilar é contínua e não exis
tem fendas (Figura 1.138). Para entrar no cérebro, o fármaco precisa pas
saratravés das células endoteliais dos capilares do SNC ou sertranspor
tado ativamente. Porexemplo, otransportadorespecífico de aminoácidos
neutros transporta levodopa para o interior do cérebro. Em contraste, fár
macos lipossolúveis entram facilmente no SNC, pois podem se dissolver
na membrana das células endoteliais. Fármacos ionizados ou polares em
geral fracassam em entrar no cérebro, pois são incapazes de atravessar
as células endoteliais do SNC, que não apresentam junção com fendas.
Essas células firmemente justapostas formam junções estreitadas que
constituem a barreira hematencefática.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-20-320.jpg)


![plasma. Se oVd é elevado, a maiorparte do fármaco está no espaço
extraplasmático e indisponível para os órgãos excretores. Portanto,
qualquer fator que aumente oVd pode levar a um aumento na meia
-vida e prolongar a duração de ação do fármaco. (Nota: um valor
de Vd excepcionalmente elevado indica considerável sequestro do
fármaco em algum órgão ou compartimento do organismo.)
V. DEPURAÇÃO DE FÁRMACOS POR MEIO DA
-
BIOTRANSFORMAÇAO
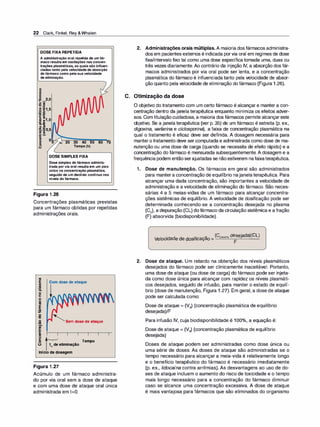
Logo que o fármaco entra no organismo inicia-se o processo de eliminação,
que envolve três vias principais:1) a biotransformação hepática; 2) a elimina
ção na bile; e 3) a eliminação na urina. Juntos, estes processos de elimina
ção fazem cair exponencialmente a concentração do fármaco no plasma. Ou
seja, a cada unidade de tempo, uma fração constante de fármaco presente é
eliminada na unidade de tempo (Figura 1.1SA). A maioria dos fármacos é eli
minada de acordo com uma cinética de primeira ordem, embora alguns, como
o ácido acetilsalicílico em altas doses, seja eliminado de acordo com cinética
de ordem zero ou não linear. A biotransformação gera produtos com maior
polaridade que facilita a eliminação. A depuração (clearance -
CL) estima a
quantia de fármaco depurada do organismo por unidade de tempo. A CL total
é uma estimativa que reflete todos os mecanismos de eliminação do fármaco
e é calculada por:
CL = 0,693 X Vjt112
onde ty2 é a meia-vida de eliminação do fármaco, Vd é ovolume de distribuição
aparente, e 0,693 é a constante log natural. A meia-vida do fármaco é usada
com frequência para mensurar a CL do fármaco porque, para vários fárma
cos, Vd é uma constante.
A. Cinética da biotransformação
1 . Cinética de primeira ordem. A transformação metabólica dos fár
macos é catalisada por enzimas, e a maioria das reações obedece
à cinética de Michaelis-Menten.3
v = velocidade de biotransformação do fármaco = �
:á:�
g
�
Na maioria das situações clínicas, a concentração do fármaco, [C],
é muito menor do que a constante de Michaelis, Km, e a equação se
reduz para:
v = velocidade de biotransformação do fármaco = Vm��C]
3Ver Bioquímica /lustrada, 4ª edição, Artmed Editora, para uma discussão
sobre cinética Michaelis-Menten.
ê.
o
-
ai
E
UI
ai
-
a.
Farmacologia Ilustrada 13
Fasede Fasede
distribuição eliminação
,._,.__ �
-
-
-
........._
_:.,_
_
_
_
4
1
g 2
A maioria dosfármacos
apresenta diminuição
exponencial naconcen
traçãoemfunção do
tempo, duranteafase
deeliminação.
o
•Ili
�
-=
e
8
e§
1
º �
=::::::::::
�
o 1 2 3 4
J.
Tempo
! - Injeçãorápidadofármaco
m
Extrapolação até o
tempo "O" forneceo C0,
ovalorhipotético de
concentração dofárma
co previstosea distri
buição fossealcançada
instantaneamente.
ai
E
UI
ai
-
a.
4
3
2
o
e Co=1
o
lCll
""
f!
'E
0,5
0,4
0,3
1
1
1
1
CI)
u
e
o
o 0,2 :'-ri
0,1
o 1
'
t
Injeçãorápida
dofármaco
t112 1
1
1
1
1
2 3
Tempo
A meia-vida(tempo necessário para
reduzirà metade a concentração do
fármaco no plasma) é igual a 0,69VJCL.
4
Figura 1 .15
Concentrações do fármaco no plasma
após uma injeção única de um fár
maco no tempo = O. A. Os dados de
concentração foram lançados em uma
escala linear. B. Os dados de concen
tração foram lançados em uma escala
logarítmica.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-23-320.jpg)
![14 Clark, Finkel, Rey &Whalen
Poucosfármacos, como ácido acetilsali
cílico, etanole fenitoína,têm dosagens
muito elevadas. Por isso, a concentração
dofármaco no plasma é muito maior do
queo Km, eassuas biotransformações
são de ordem zero, isto é, constantee
independenteda dosedofármaco.
Paraa maioria dosfármacos, a concen
tração no plasmaé muito menor do que
o Km ea eliminaçãoé deprimeiraordem,
istoé, proporcional à dosedofármaco.
Figura 1.16
Efeitos da dose do fármaco na veloci
dade da sua biotransformação.
Fármaco
Isto é, a velocidade de biotransformação do fármaco é diretamente
proporcional à concentração do fármaco livre, e é observada uma
cinética de primeira ordem (Figura 1.16). Isso indica que uma fra
ção constante do fármaco é biotransformada por unidade de tem
po (isto é, a cada meia-vida, a concentração se reduz em 50o/
o). A
cinética de primeira ordem algumas vezes é referida clinicamente
como cinética linear.
2. Cinética de ordem zero. Com poucos fármacos, como o ácido acetil
salicílico, o etanole a fenitoí
na, as doses são muito grandes. Por isso,
[C] é muito maiordo que Km, e a equação de velocidade se torna:
V = velocidade de biotransformação do fármaco = Vm[c�C]= Vmáx
A enzima é saturada pela concentração elevada de fármaco livre, e a
velocidade da biotransformação permanece constante notempo. Isso
é denominado cinética de ordem zero (também referida clinicamente
como cinética não linear). Uma quantidade constante do fármaco é
biotransformada por unidade de tempo e a velocidade de eliminação
é constante e não depende da concentração do fármaco.
B. Reações da biotransformação de fármaco
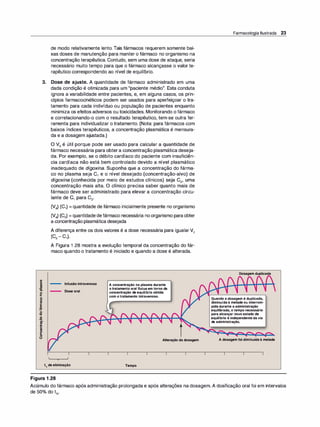
Os rins não conseguem eliminar os fármacos lipofílicos de modo eficien
te, pois eles facilmente atravessam as membranas celulares e são reab
sorvidos nos túbulos contorcidos distais. Por isso, os fármacos lipossolú
veis primeiro devem serbiotransformados no fígado em substâncias mais
polares (hidrofílicas) usando dois grupos gerais de reações, denomina
dos Fase 1 e Fase li (Figura 1 .17).
1. Fase 1. As reações de Fase 1 convertem moléculas lipofílicas em
moléculas mais polares, introduzindo ou desmascarando um grupo
funcional polar, como -OH ou -NH2• A biotransformação de Fase 1
pode aumentar, diminuir ou deixar inalterada a atividade farmacoló
gica do fármaco.
a. Reações de Fase 1 utilizando o sistema P450. As reações de
Fase 1 envolvidas com mais frequência na biotransformação de
fármacos são catalisadas pelo sistema citocromo P450 (tam
bém denominado oxidases microssomais de função mista):
Fármaco + 02 + NADPH + H+ � Fármacomoditicado + H20 + NADP+
Oxidação,
redução,
e/ou hidrólise
Algunsfármacosentram
diretamente na Fase li
de biotransformação.
� Produtos de
conjugação
Faseli
!li
Após aFase1, ofármaco pode serativado, perma
necer inalteradoou, com maisfrequência, inativado.
O fármacoconjugado
geralmenteé inativo.
Figura 1.17
Biotransformação dos fármacos.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-24-320.jpg)








![26 Clark, Finkel, Rey &Whalen
ft Canaisiônicos
W disparadosporligantes
Exemplo:
Receptorescolinérgicos
nicotínicos
,
lons
Esse conceito está estreitamente relacionado com a formação de comple
xos entreenzimas esubstratos ou antígenos eanticorpos. Essas interações
têm vários aspectos comuns, provavelmente a mais notável seja a especifi
cidade do receptor por um determinado ligante. Contudo, o receptor não só
tem a habilidade de reconhecer o ligante, mas também acopla ou transduz
essa ligação em uma resposta causando uma alteração conformacional ou
um efeito bioquímico. A maioria dos receptores é denominada para indicar
o tipo de fármaco que melhor interage com ele. Por exemplo, o receptor da
histamina é denominado receptor histamínico. Embora muito deste capítulo
esteja focado na interação dos fármacos com os receptores específicos, é
importante estar ciente de que nem todos os fármacos exercem seus efei
tos interagindo com um receptor. Os antiácidos, por exemplo, neutralizam
quimicamente o excesso de ácido gástrico, reduzindo os sintomas de azia.
B. Estados receptores
Classicamente se pensava que a fixação de um ligante modificava os re
ceptores de um estado inativo (R) para um ativado (R*). O receptor ativado
então interagia com moléculas efetoras intermediárias provocando o efeito
biológico. Este modelo é um esquema simples e intuitivo e é usado nas
ilustrações deste capítulo. Informações mais recentes sugerem que os re
ceptores existem em dois estados, no mínimo, o inativo (R) e o ativo (R*)
que estão em equilíbrio reversível entre si. Na ausência do agonista, o R*
representa tipicamente uma pequenafraçãodototal da populaçãode recep
tores (ou seja, oequilíbriofavorece o estado inativo). Fármacos que ocupam
o receptor podem estabilizar o receptor num certo estado conformacional.
Alguns fármacos podem causardeslocamentos similares noequilíbrio entre
R e R* como um ligante endógeno. Porexemplo, fármacos que atuam como
agonistas se ligam ao estado ativo dos receptores e, assim, rapidamente
deslocam o equilíbrio de R para R*. Outros fármacos podem induzir altera
ções que podem ser diferentes dos ligantes endógenos. Essas alterações
tornam os receptores menos funcionais ou não funcionais.
� �ecept?resacoplados
l:il aprote1naG I'!! Rece�toresligados
� aenz1mas Receptores
intracelulares
Exemplo:
Adrenorreceptoresa ep Exemplo:
Receptoresdeinsulina
Oi
n
R�R-P
l
I
l
I
l
I
Exemplo:
Receptoresesteroides
l l
r Ãmnm
I I l I I
-
,
-v-
J t;
Alteraçõesnopotencialde Fosforilaçãodeproteínas Fosforilaçãodeproteínas
J
doreceptor Proteínasealteração
daexpressãogênica
membranaouconcentração
iônicanointeriordacélula :::]
EFEITOS INTRACELULARES
Figura 2.2
Mecanismos de sinalizaçãotransmembrana. A. O ligante se une a domínios extracelulares do canal estimulado por ligan
te. B. O ligante se une a um domínio no receptor transmembrana, que está acoplado à proteína G. C. O ligante se une
ao domínio extracelular de um receptor que ativa uma enzima quinase. D. O ligante lipossolúvel difunde-se através da
membrana para interagir com seu receptor intracelular. R = proteína inativa.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-36-320.jpg)



![30 Clark, Finkel, Rey &Whalen
l!I
A administração repetidade
um agonista (como a epinefrina)
em curto período detempo
resulta na diminuição da
resposta dacélula.
l
:i
a:
Injeção repetidado fármaco
Após um período de repouso,
aadministração dofármaco
resultaem uma resposta
de intensidadeoriginal.
Figura 2.6
Dessensibilização de receptores.
rJ
100 Fármaco A
o
E
·-
·
=
E
o
-
·
-
GI
ãi 50
o
"O
E
&
m
CEso
e
/'1
8
...
o o 1
Q.
.,.
Ili.
gem, exigem um tempo finito (período de repouso) após a estimula
ção antes de poderem ser ativados novamente. Durante essa fase
de recuperação, eles são "refratários" ou "não responsivos".
-
RELAÇOES DOSE-RESPOSTA
O agonista é definido como o fármaco que pode se ligar ao receptor e provo
car um efeito biológico. Um agonista em geral mimetiza a ação de um ligante
endógeno original no seu receptor, como a norepinefrina nos receptores 131
cardíacos. A intensidade do efeito depende da concentração do fármaco no
local do receptor que, por sua vez, é determinada pela dose do fármaco ad
ministrado e por fatores característicos do perfil farmacocinético do fármaco,
velocidades de absorção, distribuição e biotransformação.
A. Relações dose-resposta graduais
À medida que a concentração de um fármaco aumenta, a intensidade do
seu efeito farmacológicotambém aumenta.A respostaé um efeito gradual,
ou seja, ela é contínua e graduada. Lançando a intensidade da resposta
contra as doses de um fármaco, obtém-se um gráfico que apresenta o for
mato geral apresentado na Figura 2.7A. A curva pode ser descrita como
uma hipérbole retangular- uma curva muito familiar em biologia, pois pode
ser aplicada a diversos eventos biológicos, como a ligação de fármacos,
a atividade enzimática e as respostas aos agentes farmacológicos. Duas
propriedades importantes dosfármacos, potência e eficácia, podem serde
terminadas nas curvas dose-resposta graduadas.
1 . Potência. A primeira propriedade é a potência, uma medida da
quantidade de fármaco necessária para produzir um efeito de uma
dada intensidade.A concentração de fármaco que produz um efeito,
que é igual a 50°
/
o do máximo, é usada para determinar a potência;
comumente isso é designado como CE50•Na Figura 2.7, a CE50dos
fármacos A e B estão indicadas. O fármacoA é mais potente do que
o B porque menorquantidade de fármaco A é necessário para obter
50°
/
o do efeito. Assim, as preparações terapêuticas dos fármacos
refletem a potência. Por exemplo, candesartano e irbesartano são
bloqueadores de receptores da angiotensina, que são usados indi-
m
0 100
E
·-
·=
Fármaco B E
o Fármaco B
-
·
-
GI
ãi
.g 50 -
-
-
-
-
-
-
-
E
&
m
-
e CEso
8
...
o
Q. o
[Fármaco] log [Fármaco]
Figura 2.7
A CEsoé a concentração dofármaco
que produz uma resposta igual a 50°/o
da resposta máxima.
A potência dofármaco podeser
comparada usando a CEso: quanto
menora CEso, mais potenteé ofármaco.
O efeito da dose na intensidade da resposta farmacológica. A. Gráfico em escala linear. B. Lançamento semilogarítmico
dos mesmos dados. CE50= dose do fármaco que provoca 50°
/
o da resposta máxima.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-40-320.jpg)
![vidualmente ou em associação para o tratamento da hipertensão.
O candesartano é mais potente que o irbesartano, pois a faixa de
doses do candesartano é de 4 a 32 mg, comparado com a faixa de
75 a 300 mg do irbesartano. O candesartano seria o fármaco A e
o irbesartano o fármaco B na Figura 2.7. Gráficos semilogarítmi
cos são empregados com frequência, pois a faixa das doses (ou
concentrações) pode ter uma distribuição muito ampla. Lançando o
log da concentração, a faixa completa de doses pode ser lançada.
Como apresentado na Figura 2.78, as curvas assumem a forma
sigmoide, o que simplifica a interpretação da dose e resposta.
2. Eficácia. A segunda propriedade que pode ser determinada das cur
vas dose-resposta é a eficácia do fármaco. Ela significa a habilidade
do fármaco de provocar a resposta farmacológica quando interage
com um receptor.A eficácia depende do número de complexos fárma
correceptorformados eda eficiênciado acoplamentodesde a ativação
do receptor até a resposta celular. Em analogia com a velocidade má
xima das reações catalisadas por enzimas, a resposta máxima (Emáx)
ou eficácia é mais importante do que a potência do fármaco. Um fár
maco com maior eficácia é terapeuticamente mais benéfico do que
um que é mais potente. A eficácia máxima de um fármaco considera
que todos os receptoresestão ocupados pelofármaco e não se obterá
aumento na resposta se mais fármaco for administrado. Este conceito
só éválido se não existem"receptores de reserva" (ver p. 29).A Figura
2.8 mostra a resposta afármacos de diferentes potências e eficácias.
B. Efeito da concentração do fármaco nas ligações com o
receptor
A relação quantitativa entre a concentração do fármaco e a ocupação
dos receptores aplica a lei de ação das massas à cinética de ligação do
fármaco com as moléculas receptoras:
Fármaco + Receptor � Complexo fármaco-receptor--7 Efeito biológico
Admitindo que a ligação de uma molécula não altera a ligação de mo
léculas subsequentes e aplicando a lei de ação das massas, pode-se
expressar matematicamente a relação entre a porcentagem (ou a fração)
de receptores ocupados e a concentração do fármaco:
[DR] _
[Rt]
[D]
Kd + [D]
(1)
em que [D] = a concentração do fármaco livre; [DR] = a concentração do
fármaco ligado; [R1] = a concentração total de receptores, que é igual à
soma dos receptores ocupados e dos receptores não ocupados (livres);
e Kd= a constante de dissociação de equilíbrio para o fármaco do recep
tor. O valor Kd pode ser usado para determinar a afinidade do fármaco
pelo seu receptor. A afinidade descreve a força da interação (ligação)
entre o ligante e o seu receptor. Quanto maior o valor de Kd, mais fraca
é a interação e menor a afinidade. O fenômeno inverso ocorre quando o
fármaco tem um valorde Kd baixo.A fixação do ligante ao receptor é forte
e a afinidade elevada. A equação (1) define a curva que tem a forma de
uma hipérbole retangular (Figura 2.9A). À medida que a concentração do
fármaco livre aumenta, a relação entre a concentração do receptor ligado
e a concentração dos receptorestotais se aproxima da unidade.A ligação
Farmacologia Ilustrada 31
OfármacoA é mais
potentedoqueo
fármacoB,mastem
amesmaeficácia.
ofármacoc apre
sentamenorpo
tênciaemenor
eficáciadoqueos
fármacosAeB.
100
o
.Si
CJI
•O
õ50 - - - -
:s
o
-
·-
itl
Fármaco Fármaco
1 A - -
B
1
1
1- - - - - - - - - - -
1 1
á
1 _/ 1 rmaco
, 1 : e
• 1
o
Logdaconcentraçãodefármaco
t
CE50
do
fármacoA
Figura 2.8
t
CE50
do
fármacoB
t
CE50
do
fármacoe
Curvas dose-resposta típicas para fár
macos que mostram diferenças em
potência e eficácia. (CE50 = dose do
fármaco que provoca 50o/
o da resposta
máxima.)
'iii' 1
o ......
"O Ul
m ·
-
e. .l!I
� g
o Ul
e � o,5
g e.
e. QI
� �
a: ......
......
Figura 2.9
Dose
Logdose
O efeito da dose na intensidade da liga
ção do fármaco.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-41-320.jpg)
![32 Clark, Finkel, Rey &Whalen
O agonista total provoca
a ativação total do receptor
nas concentrações
elevadas do fármaco.
A ligação do agonista parcial
resulta na ativação de menos
de 100'Y
o, mesmo em concen
trações muito elevadas.
100
...
s 75
8
I!!
o
"O
Agonistaparcial
.g 50
�
·
-
>
·-
êt
25
Logdaconcentraçãodofármaco
O agonista inverso provoca
uma resposta aquém da linha
de base mensurada na
ausência de fármaco.
Neste exemplo, cerca de 12% dos
receptores têm atividade
constitutiva na ausência do
agonista.
Figura 2.10
Efeitos dos agonistas total, parcial e in
verso na atividade do receptor.
do fármaco ao seu receptor inicia eventos que, no final, levam à resposta
biológica mensurável. Assim, não é surpresa que as curvas mostradas
na Figura 2.9 e aquelas que representam a relação entre dose e efeito
(vistas na Figura 2.7) sejam similares.
C. Relações da ligação do fármaco com o efeito farmacológico
O modelo matemático que descreve a concentração dofármaco e a ligação
ao receptor pode ser aplicado à dose (concentração do fármaco) e à res
posta (ou efeito), desde que as seguintes premissas sejam atendidas: 1) o
tamanho da resposta é proporcional à quantidade de receptores ligados ou
ocupados; 2) o Emáx ocorre quando todos os receptores estão ocupados; e
3) a ligação dofármaco ao receptor não exibe cooperatividade. Nesse caso,
[E]
[Emáx1
[D]
- -
�
-
Kd + [D]
(2)
em que [E] = o efeito do fármaco na concentração [D] e [Emáxl = o efeito
máximo do fármaco.
IV. AGONISTAS
Um agonista se liga ao receptor e produz uma resposta biológica. Um ago
nista pode mimetizar a resposta do ligante endógeno no receptor ou pode
provocar uma resposta diferente deste receptor e seu mecanismo.
A. Agonistas totais
Se um fármaco se liga a um receptor e produz a resposta biológica máxi
ma que mimetiza a resposta do ligante endógeno, ele é denominado ago
nista total (Figura 2.1 O). Os receptores existem nos estados conformacio
nais ativo e inativoque estão em equilíbrio reversível entre si. Os fármacos
que ocupam o receptor podem estabilizá-lo em um determinado estado
conformacional. Assim, outra definição de agonista é um fármaco que se
liga ao receptor estabilizando-o no seu estado conformacional ativo. Por
exemplo, a fenilefrina é um agonista nos adrenoceptores a, porque provo
ca efeitos que se assemelham à ação do ligante endógeno norepinefrina.
Após ligar-se ao adrenoceptor a, na membrana do músculo liso vascular,
a fenilefrina estabiliza o receptor em seu estado ativo. Isso mobiliza Ca
2
+
intracelular, causando interação dos filamentos de actina e miosina. O en
curtamento das células musculares diminui o diâmetrodas arteríolas, cau
sando um aumento na resistência ao fluxo sanguíneo, através dos vasos.
A pressão arterial aumenta para manter esse fluxo. Como esta descrição
breve ilustra, um agonista pode tervários efeitos que podem ser medidos,
incluindo ações em moléculas intracelulares, em células, em tecidos e no
organismo inteiro. Todas essas ações são atribuídas à interação da mo
lécula do fármaco com a molécula receptora. Em geral, um agonista total
tem elevada afinidade pelo seu receptor e boa eficácia.
B. Agonistas parciais
Os agonistas parciais têm eficácia (atividade intrínseca) maior do que
zero, mas menordo que a de um agonistatotal (Figura 2.10). Mesmo que
todos os receptores sejam ocupados, os agonistas parciais não conse
guem produzir um Emáx da mesma amplitude de um agonista total. Entre
tanto, o agonista parcial pode ter uma afinidade que é maior, menor ou
equivalente à do agonistatotal. A característicasingular dessesfármacos](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-42-320.jpg)


![,
A. lndice terapêutico
O índice terapêutico (IT) de um fármaco é a relação da dose que produz
toxicidade com a dose que produz o efeito eficaz ou clinicamente deseja
do em uma população de indivíduos:
Índice terapêutico = DT50 1 DE50
em que DT50 = a dose do fármaco que produz efeito tóxico em metade da
população, e DE50 = a dose do fármaco que produz uma resposta tera
pêutica ou desejada na metade da população. O índice terapêutico é uma
mensuração da segurança do fármaco, pois um valor elevado indica uma
grande margem entre as doses que são efetivas e as que são tóxicas.
B. Determinação do índice terapêutico
O índice terapêutico é determinado pela mensuração da frequência da
resposta desejada e da resposta tóxica em várias doses do fármaco. Por
convenção, as doses que produzem o efeito terapêutico (DE50) e o efei
to tóxico em 50°
/
o (DT50) da população são empregadas. Em humanos,
o índice terapêutico é determinado pelo uso de triagem do fármaco e
pela experiência clínica acumulada. Em geral, eles revelam uma faixa de
doses eficazes e uma faixa distinta (algumas vezes, com sobreposição)
de doses tóxicas. Embora alguns fármacos tenham índices terapêuticos
pequenos, eles são usados rotineiramente para tratar certas doenças.
Várias doenças letais, como linfomade Hodgkin, são tratadas com fárma
cos que têm índice terapêutico pequeno; mas, porexemplo, o tratamento
de uma cefaleia simples com fármaco de índice terapêutico pequeno é
inaceitável. A Figura 2.13 mostra as respostas à varfari
na, um anticoa
gulante oral com índice terapêutico pequeno, e à penicilina, um fármaco
antimicrobiano com índice terapêutico amplo.
1. Varfarina (exemplo de fármaco com índice terapêutico peque
no). À medida que a dose de varfarina aumenta, uma maior fração
dos pacientes responde (para essefármaco, a resposta desejada é o
aumento de duas a três vezes na relação normalizada internacional
[INR]), até quefinalmente todos os pacientes respondem (ver Figura
2.13A). Contudo, nas doses mais elevadas de varfarina, ocorre uma
resposta tóxica, ou seja, um elevado grau de anticoagulação que
resulta em hemorragia. (Nota: quando o índice terapêutico é baixo,
é possível ter uma faixa de concentrações em que a resposta eficaz
e a tóxica se sobrepõem. Isto é, alguns pacientes têm hemorragia, e
outros alcançam o prolongamento desejado de duas a três vezes no
INR.) A variação na resposta do paciente é, portanto, mais provável
de ocorrer com um fármaco que tem índice terapêutico baixo, pois
as concentrações eficazes e tóxicas são similares. Fármacos com
índices terapêuticos pequenos - ou seja, fármacos para os quais a
dose é crucialmente importante - são aqueles cuja biodisponibilida
de altera de modo crítico o efeito terapêutico (ver p. 8).
2. Penicilina (exemplo de fármaco com índice terapêutico amplo).
Para fármacos como a penicilina (ver Figura 2.138), é seguro e co
mum administrar doses em excesso (com frequência, com excesso
de 1O vezes) daquela que é a minimamente necessária para alcan
çar a resposta desejada. Nesse caso, a biodisponibilidade não alte
ra criticamente os efeitos terapêuticos ou clínicos.
cn
J!!
ii100
·
-
u
!
CP
Farmacologia Ilustrada 35
Varfarina: índice
terapêutico pequeno
Janela
terapêutica
,---A--..
'O
50 Efe�o
E terapêutico Efeito
& desej�do adverso
� 1
(indesejado)
� º �
�
�
----
�
�
�
�
o
a.
cn
� 100
CP
'ij
li
-8
E50
i
e
B
ê5 o
a.
Logconcentraçãodo
fármaconoplasma
(unidadesarbitrárias)
Penicilina: índice
terapêutico amplo
Janela
terapêutica
Efeito
terapêutico Efeito
desejado adverso
Logconcentraçãodo
fármaconoplasma
(unidadesarbitrárias)
(indesejado
Figura 2.13
Porcentagem cumulativa de pacientes
que respondem aos níveis plasmáticos
de varfarina e benzi/penicilina.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-45-320.jpg)





![(Nota: em cada caso, o arco reflexo do SNA compreende um ramo
sensorial [aferente] e um ramo motor [eferente ou efetor]).
2. Emoções e o SNA. Estímulos que provocam sensações fortes,
como raiva, medo ou prazer, podem modificar a atividade do SNA.
F. Inervação pelo sistema nervoso autônomo
1 . Inervação dupla. A maioria dos órgãos do organismo é inervada
por ambas as divisões do SNA. Assim, a inervação parassimpáti
ca vagai diminui a frequência cardíaca, e a inervação simpática a
aumenta. Apesar dessa inervação dual, um sistema em geral pre
domina no controle da atividade de um dado órgão. Por exemplo,
no coração, o nervo vago é o fator predominante no controle da
frequência. Esse tipo de antagonismo é dinâmico e tem ajuste fino a
cada instante, visando ao controle homeostático da função orgâni
ca. A atividade de um sistema representa a integração da influência
das duas divisões.
2. Órgãos que só recebem a inervação simpática. Embora a maioria
dos tecidos receba inervação dual, alguns órgãos efetores, como a
medulasuprarrenal, os rins, os músculos piloeretores e as glândulas
sudoríparas, recebem somente inervação do sistema simpático. O
controle da pressão arterial também é uma atividade principalmente
simpática, sem participação significativa do sistema parassimpático.
G. Sistema nervoso somático
O sistema nervoso somático eferente difere do sistema autônomo pelo
fato de um único neurônio motor mielinizado, originado no SNC, ir direta
mente ao músculo esquelético sem a intermediação de gânglios. Como
já salientado, o sistema nervoso somático está sob controle voluntário, e
o autônomo é um sistema involuntário. Em geral, as respostas na divisão
somática são mais velozes do que as do SNA.
H. Resumo das diferenças entre os nervos simpáticos,
parassimpáticos e motores
As principais diferenças na organização anatômica dos neurônios levam
a variações nas funções de cada divisão (Figura 3.6). O sistema nervoso
simpático é amplamente distribuído, inerva praticamente todos os siste
mas efetores do organismo. Em contraste, a distribuição dadivisão paras-
Farmacologia Ilustrada 41
D INFORMAÇÃO AFERENTE
Impulsossensoriaisdeorigemvisceral:
• Quedanapressãoarterial.
• Menorestiramentodos
barorreceptoresnoarcoaórtico.
• Menorfrequênciadosimpulsos
aferentesparaobulbo(troncocerebral).
fl RESPOSTA REFLEXA
Impulsoseferentesreflexosatravés
dosistemanervosoautônomocausam:
• Inibiçãodadivisãoparassimpática
eativaçãodadivisãosimpática.
• Aumentodaresistênciaperiféricae
dodébitocardíaco.
• Aumentoda pressãoarterial.
Figura 3.5
Arco reflexo barorreceptor responde à
diminuição da pressão arterial.
SIMPÁTICO PARASSIMPÁTICO •
Origem
Comprimento das fibras
Localização dos gânglios
Ramificação das fibras pré-ganglionares
Distribuição
Tipos de respostas
Figura 3.6
Região torácica e lombar da medula
espinal (toracolombar)
Pré-ganglionares curtas
Pós-ganglionares longas
Próximos à medula
Extensa
Ampla
Difusas
Características dos sistemas nervosos simpático e parassimpático.
•
Area cerebral e sacral da medula
espinal (craniossacral)
Pré-ganglionares longas
Pós-ganglioinares curtas
Próximo ou no interior do órgão efetor
Mínima
Limitada
Discretas](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-51-320.jpg)

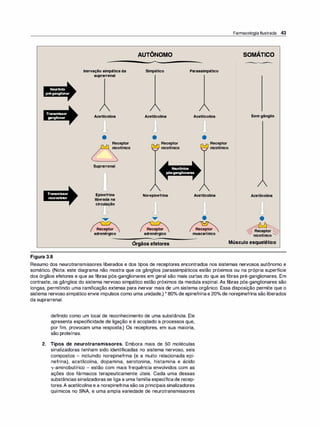

![b. Norepinefrina e epinefrina. Quando a norepinefrina ou a epi
nefrinaé o transmissor, a fibra é denominada adrenérgica (adre
nalina é outro nome da epinefrina). No sistema simpático, a no
repinefrina intermedeia a transmissão dos impulsos dos nervos
pós-ganglionares autônomos para o órgão efetor. A norepinefri
na e os receptores adrenérgicos são discutidos nos Capítulos 6
e 7. Um resumo dos neuromediadores liberados e do tipo de re
ceptores do sistema nervoso periférico é apresentado na Figu
ra 3.9. (Nota: poucas fibras simpáticas, como as envolvidas na
sudoração, são colinérgicas, e, para simplificação, elas não são
representadas na figura. O músculo liso renal pós-ganglionar é
inervado pela dopamina.)
IV. SISTEMAS DE SEGUNDO MENSAGEIRO NA
RESPOSTA INTRACELULAR
A ligação dos sinalizadores químicos aos receptores ativa processos enzi
máticos no interior da membrana celular. No final, esses processos resultam
em uma resposta celular, como a fosforilação de proteínas intracelulares ou
alterações na condutividade de canais iônicos. O neurotransmissor pode ser
imaginado como um sinal; e o receptor, como o detector do sinal e transdu
tor. Moléculas segundas mensageiras, produzidas em resposta à ligação do
neurotransmissor, traduzem o sinal extracelular em uma resposta que pode
ser propagada mais adiante ou amplificada no interior da célula. Cada com
ponente serve como um elo na comunicação entre eventos extracelulares e
alterações químicas no interior da célula (ver Capítulo 2).
A. Receptores de membrana que afetam a permeabilidade
.- .
1on1ca
Os receptores de neurotransmissores são proteínas de membrana que
disponibilizam o local de ligação que reconhece e responde à molécula
neurotransmissora. Alguns receptores, como os receptores pós-sinápti
cos dos nervos e músculos, são ligados diretamente a canais iônicos de
membrana; assim, a ligação do neurotransmissor ocorre de modo rápido
(em fração de milissegundos) e afeta diretamente a permeabilidade iô
nica (Figura 3.1OA). (Nota: o efeito da acetilcolina nesses canais iônicos
disparados porsubstância química é discutido na p. 27.)
B. Regulação envolvendo moléculas segundas mensageiras
Vários receptores não são acoplados diretamente a canais iônicos. Nes
ses casos, o receptor sinaliza o reconhecimento da ligação de um neu
rotransmissor iniciando uma série de reações, que no final resulta em
uma resposta intracelular específica. Moléculas segundas mensagei
ras - assim denominadas porque intervêm entre a mensagem inicial (o
neurotransmissor ou hormônio) e o efeito final na célula - são parte de
uma cascata de eventos que traduzem a ligação do neurotransmissorem
uma resposta celular, em geral, por meio da intervenção de uma proteína
G. Os dois segundos mensageiros mais amplamente reconhecidos são
os sistemas adenililciclase e cálcio-fosfatidilinositol (Figura 3.108 e C).
(Nota: G5 é uma proteína envolvida na ativação da adenililciclase, e Gq
é uma subunidade que ativa a fosfolipase C para liberar diacilglicerol e
trifosfato de inositol [ver p. 27].)
Farmacologia Ilustrada 45
ft Recep�or!s�copiados
W acanais1on1cos
Neurotransmissor
Espaço
extracelular
Membrana
celular
Membranacelular
Citosol c1-
�
Alteraçõesnopotencialde
membranaouconcentração
iônicanointeriordacélula
r:'I Receptoresacoplados
l:il àadenililciclase
Q Hormônioou
� neurotransmissor
Adenililciclase
ativa
AMPc+ PPi
�
Fosforilaçãodeproteínas
f':! Receptoresacopladosao
� diacilgliceroleao
trifosfatodeinositol
ProteínaGq
Diacilglicerol
�
FosfolipaseC
Trifosfato
deinositol
Fosforilaçãodeproteína
eaumentodoCa2+intracelular
Figura 3.10
Três mecanismos pelos quais a ligação
com o neurotransmissor leva a um efeito
biológico.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-55-320.jpg)

![. , •
o 1 ner ICOS
1. RESUMO
Os fármacos que afetam o sistema nervoso autônomo (SNA) são divididos
em dois grupos, de acordo com o tipo de neurônio envolvido nos seus me
canismos de ação. Os fármacos colinérgicos (descritos neste capítulo e no
seguinte) atuam em receptores que são ativados pela acetilcolina (ACh), e os
fármacos adrenérgicos (discutidos nos Capítulos 6 e 7) atuam em receptores
que são estimulados pela norepinefrina ou epinefrina. Os fármacos colinérgi
cos e adrenérgicos atuam estimulando ou bloqueando receptores do SNA. A
Figura 4.1 resume os agonistas colinérgicos discutidos neste capítulo.
li. O NEURÔNIO COLINÉRGICO
A fibra pré-ganglionar que termina na suprarrenal, o gânglio autônomo (tanto
parassimpático como simpático) e as fibras pós-ganglionares da divisão pa
rassimpática usam acetilcolina como neurotransmissor (Figura 4.2). A divisão
pós-ganglionar simpática das glândulas sudoríparas também usa ACh. Além
disso, neurônios colinérgicos inervam os músculos do sistema somático e
também desempenham função importante no SNC. (Nota: pacientes com a
doença de Alzheimer têm perda significativa dos neurônios colinérgicos no
lobo temporal e no córtex entorrinal. A maioria dos fármacos disponíveis para
tratar essa doença são inibidores da acetilcolinesterase [ver p. 108].)
A. A neurotransmissão nos neurônios colinérgicos
A neurotransmissão nos neurônios colinérgicos envolve seis etapas sequen
ciais: 1) síntese, 2) armazenamento, 3) liberação, 4) ligação da ACh ao recep
tor, 5) degradação do neurotransmissor na fenda sináptica (ou seja, o espaço
entre os terminais nervosos e os receptores adjacentes localizados nos ner
vos ou órgãos efetores), e 6) reciclagem de colina e acetato (Figura 4.3).
1. Síntese de acetilcolina. A colina é transportada do líquido extrace
lular para o citoplasma do neurônio colinérgico por um sistema car
regador dependente de energia que cotransporta sódio e pode ser
AÇÃO DIRETA
Acetilcolina
Betanecol
Carbacol
Cev
imelina
Pilocarpina
AÇÃO INDIRETA (reversíveis)
Ambenônio
Donepezila
Galantamina
Neostigmina
Fisostigmina
Piridostigmina
Rivastigmina
Tacri
na
AÇÃO INDIRETA (irreversíveis)
Ecotiofato
REATIVADOR DE
ACETILCOLINESTERASE
Pralidoxima
Figura 4.1
Resumo dos agonistas colinérgicos.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-57-320.jpg)




![52 Clark, Finkel, Rey &Whalen
Diarreia
Diaforese
Miose
Náusea
- -
v=�
Emergência --
urinária
Figura 4.6
Alguns efeitos adversos observados
com os agonistas colinérgicos.
A. Acetilcolina
A acetilcolina é um composto amônia quaternário que não consegue
penetrar membranas. Embora seja o neurotransmissor de nervos paras
simpáticos e somáticos, bem como dos gânglios autônomos, não tem
importância terapêutica devido à sua multiplicidade de ações (provocan
do efeitos difusos) e à sua rápida inativação pelas colinesterases. A ACh
tem atividade muscarínica e nicotínica. Suas ações são citadas a seguir:
1. Diminuição da frequência e do débito cardíaco. As ações daACh
no coração mimetizam os efeitos da estimulação vagai. Por exem
plo, se injetada IV, a ACh produz uma breve redução na frequência
cardíaca (cronotropismo negativo) e novolume sistólico como resul
tado da redução dafrequênciade descargas no nó sinoatrial (NSA).
(Nota: deve ser lembrado que a atividade vagai normal regula o co
ração pela liberação de ACh no NSA.)
2. Diminuição da pressão arterial. A injeção de ACh causavasodila
tação e diminuição da pressão sanguínea por mecanismo indireto.
A ACh ativa receptores M3 situados nas células endoteliais que co
brem o músculo liso dos vasos sanguíneos. Isso resulta na produ
ção de óxido nítrico a partir de arginina.
3
(Nota: o óxido nítrico [NO]
também é denominado fator relaxante derivado do endotélio.) (Ver
p. 363 para mais informações sobre NO.) O NO então difunde-se
até as células musculares lisas dos vasos para estimular a produ
ção de proteína-quinase G, levando à hiperpolarização e ao relaxa
mento do músculo liso por meio da inibição dafosfodiesterase-3. Na
ausência da administração de fármacos colinérgicos, os receptores
vasculares não têm função conhecida, pois a ACh nunca é liberada
no sangue em quantidade significativa. A atropina bloqueia esses
receptores muscarínicos e evita que a ACh produza vasodilatação.
3. Outras ações. NoTGI, aACh aumenta a secreção salivare estimula
as secreções e a motilidade intestinal. As secreções bronquiais tam
bém são aumentadas. Notrato geniturinário, a ACh aumenta otônus
do músculo detrusor da urina, causando emissão da urina. No olho,
a ACh estimula a contração do músculo ciliar para a visão próxima e
contrai o esfíncterda pupila, causando miose (constrição acentuada
da pupila). A ACh (em solução a 1°
/
o) é instilada na câmara anterior
do olho para produzir miose durante cirurgias oftálmicas.
B. Betanecol
O betanecol é um éster carbamoila não substituído, estruturalmente re
lacionado com a ACh, na qual o acetato é substituído por carbamato e a
colina é metilada (ver Figura 4.5). Portanto, o betanecolnão é hidrolisado
pela AChE (devido à esterificação do ácido carbâmico), embora seja ina
tivado por meio de hidrólise por outras colinesterases. Ele não tem ações
nicotínicas (pela presença do grupo metila), mas apresenta forte ativida
de muscarínica. Suas principais ações são na musculatura lisa da bexiga
urinária e noTGI.Tem duração de ação de cerca de uma hora.
1. Ações. O betanecol estimula diretamente os receptores muscarí
nicos, aumentando a motilidade e o tônus intestinal. Ele também
estimula o músculo detrusor da bexiga enquanto relaxa o trígono
e os esfíncteres. Esses efeitos aumentam a pressão de micção e
diminuem a capacidade da bexiga, causando expulsão da urina.
3Ver Bioquímica /lustrada, 4ª edição, Artmed Editora, para discussão sobre
as funções do óxido nítrico.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-62-320.jpg)
![2. Aplicações terapêuticas. No tratamento urológico, o betanecolé
usado para estimular a bexiga atônica, particularmente na retenção
urinária não obstrutiva no pós-parto ou pós-operatório. O betaneco/
também pode ser usado no tratamento da atonia neurogênica, bem
como no megacólon.
3. Efeitos adversos. O betanecolcausa os efeitos da estimulação co
linérgicageneralizada (Figura 4.6). Esses incluem sudoração (diafo
rese), salivação, rubor, diminuição da pressão arterial, náuseas, dor
abdominal, diarreia e broncoespasmo. O sulfato de atropina pode
seradministrado para superaras graves respostas cardiovasculares
ou broncoconstritoras desse fármaco.
C. Carbacol (carbamilcolina)
O carbacol apresenta ações muscarínicas e nicotínicas. Ele não tem o
grupo metila presente no betanecol(ver Figura 4.5). Como o betaneco/, o
carbacolé um ésterdo ácido carbâmico e um mau substrato para aAChE
(ver Figura 4.5). Ele é biotransformado poroutras esterases, mas em uma
velocidade muito menor.
1 . Ações. O carbacoltem amplos efeitos nos sistemas cardiovascu
lar e gastrintestinal devido à sua atividade estimulante ganglionar,
podendo primeiro estimular e depois deprimir esses sistemas. Ele
pode causar liberação de epinefrina da suprarrenal por sua ação
nicotínica. lnstilado localmente no olho, o carbacol mimetiza os efei
tos da ACh, causando miose e espasmo de acomodação, no qual o
músculo ciliar permanece em um estado constante de contração.
2. Usos terapêuticos. Devido à sua alta potência, inespecificidade por
receptor e duração de ação relativamente longa, o carbaco/ raras
vezes é usado em terapêutica, exceto no olho, como fármaco mióti
co no tratamento do glaucoma, por causar contração pupilar e uma
diminuição da pressão intraocular. O início da ação miótica é de 1O a
20 minutos. A pressão intraocular fica reduzida por 4 a 8 horas.
3. Efeitos adversos. Nas doses usadas em oftalmologia, pouco ou
nenhum efeito adverso ocorre devido à sua escassa penetrabilidade
sistêmica (o carbacol é uma amina quaternária).
D. Pilocarpina
O alcaloide pilocarpina é uma amina terciária e resiste à hidrólise pela
AChE (ver Figura 4.5). Comparado com a ACh e seus derivados, a pilo
carpina é muito menos potente, mas por não possuircarga elétrica pene
tra no SNC nas dosagens terapêuticas. A pilocarpina apresenta atividade
muscarínica e é usada primariamente em oftalmologia.
1 . Ações. Aplicada localmente na córnea, a pilocarpina produz rápida
miose e contração do músculo ciliar. Quando o olho fica em miose,
também ocorre espasmo de acomodação; a visão é fixada em algu
ma distância particular, tornando impossível focalizar (Figura 4.7).
(Nota: esse é um efeito oposto ao da atropina, um bloqueador mus
carínico, no olho [ver p. 59].) A pilocarpina é um dos mais potentes
estimulantes das secreções, como suor, lágrimas e saliva (é um se
cretagogo), mas seu emprego para esses efeitos é limitado devido
à sua falta de seletividade. O fármaco é útil em promover salivação
nos pacientes com xerostomia (secura da mucosa oral) resultante
de irradiação na cabeça e no pescoço. A síndrome de Sjõgren, ca
racterizada por xerostomia e falta de lágrimas, é tratada com com-
Farmacologia Ilustrada 53
Mlose
(Contração
da pupila)
.
.
Olhotratado
compi/ocarpina
Olhonãotratado
Mldriase
(Dilatação
da pupila)
Figura 4.7
·, Olhotratado
comatropina
Ações da pilocarpina e da atropina na
íris e no músculo ciliar do olho.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-63-320.jpg)






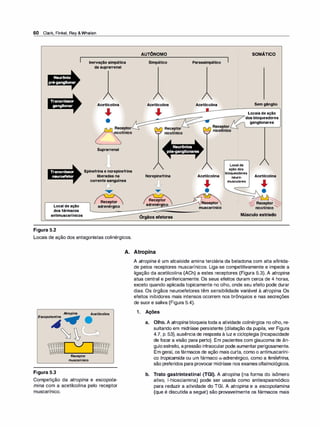
![potentes disponíveis paraesse objetivo. Emboraa motilidade do
TGI seja reduzida, a produção de ácido clorídrico não é afetada
de forma significativa. Assim, a atropina não é eficaz na cura da
úlcera péptica. (Nota: a pirenzepina [ver p. 51], um antagonista
muscarínico M,, reduz a secreção gástrica em doses que não
antagonizam outros sistemas.) Além disso, as doses de atropina
que reduzem os espasmos também diminuem a secreção sali
var, a acomodação ocular e a micção. Estes efeitos diminuem a
adesão do paciente ao tratamento com estas medicações.
c. Sistema urinário. Os fármacos tipo atropina também são em
pregados para reduzir a hipermotilidade da bexiga. Ocasional
mente ela ainda é usada na enurese (emissão involuntária da
urina) em crianças, mas os agonistas a-adrenérgicos com me
nos efeitos adversos podem ser mais eficazes.
d. Cardiovascular. A atropina produz efeitos divergentes no sistema
cardiovascular, dependendo da dose (Figura 5.4). Em doses bai
xas, o efeito predominante é a diminuição da frequência cardíaca
(bradicardia). Inicialmente atribuído à ativação central do efluxo
eferente vagai, sabe-se agora que o efeito resulta do bloqueio de
receptores M, nos neurônios inibitórios pré-juncionais (ou pré
-sinápticos), permitindo então maior liberação de ACh. Em doses
mais elevadas, a atropina bloqueia os receptores M2 no nó sino
atrial, e a frequência cardíaca aumenta modestamente. Isso em
geral requer 1 mg de atropi
na, que é uma dose mais alta do que a
administrada normalmente. A pressão arterial não é afetada, mas
em níveis tóxicos, a atropina dilata osvasos cutâneos.
e. Secreções. A atropina bloqueia as glândulas salivares, provo
cando a secura das membranas mucosas orais (xerostomia).As
glândulas salivares são muito sensíveis à atropina. As glândulas
sudoríparas e lacrimais também são afetadas. (Nota: a inibição
da secreção de suor pode causar elevação da temperatura cor
poral, o que pode ser perigoso em crianças e idosos.)
2. Usos terapêuticos
a. Oftálmico. No olho, a atropina tópica exerce efeito midriático
e cicloplégico, permitindo a mensuração de erros de refração
sem interferência da capacidade adaptativa do olho. (Nota: a
fenilefri
na ou fármacos a-adrenérgicos similares são preferidos
para a dilatação pupilar se não for necessária a cicloplegia.)
Antimuscarínicos de ação mais curta (ciclopentolato e tropica
mida) substituíram largamente a atropina devido a prolongada
midríase que ela provoca (7 a 14 dias contra 6 a 24 h com os
outros fármacos). A atropina pode induzir crise aguda de dor
ocular devido ao súbito aumento da pressão ocular em indiví
duos com glaucoma de ângulo estreito.
b. Antiespasmódico. A atropina (na forma do isômero ativo, 1-
-hiosciamina) é usada como antiespasmódico para relaxar o
TGI e a bexiga urinária.
c. Antagonista de agonistas colinérgicos. A atropina é usada
para o tratamento de doses excessivas de inseticidas inibidores
de colinesterase e de envenenamento poralguns tipos de cogu
melos (certos cogumelos contêm substâncias colinérgicas que
bloqueiam as colinesterases). Doses maciças do antagonista
podem ser necessárias durante um longo período para neutra-
11!
·-
i
QI
"CI
>10mg
Farmacologia Ilustrada 61
Alucinações e
delírio; coma
gi 5 mg
Frequênciacardíaca
rápida; palpitações;
xerostomiaacentuada;
dilatação da pupila;
algumaturbidez na
visão próxima
8
2 mg
Levedepressão
0,5 mg cardíaca; alguma
xerostomia; inibição
dasudoração
Figura 5.4
Efeitos dose-dependentes da atropina.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-71-320.jpg)




![66 Clark, Finkel, Rey &Whalen
Tempo para alcançaro bloqueio
máximo
1 Tempo para recuperar 25º/oda
�
�
- resposta máxima (min)
Atracúrio 40
2
O ci
satracúr
iosedegradaespontaneamente
no plasmaeé oúnicobloqueadorneuro
muscularnãodespolarizantecujadosagem
nãoprecisaser reduzidaempacientescom
insuficiênciarenal. Em geral,é usadoem
pacientescominsuficiênciaorgânica
múltipla, poissua biotransformaçãoé
independentedafunçãohepáticaou renal.
O cisatracúr
io é útilemventilaçãomecânica
de pacientescriticamentedoentes.
e. t • . ls
isa racur10 :
L
=:
sa
=:
=:
=:
=:
=:
=:
=:
=:
=:
=:
=:
=:
=:
=:
:
Vagolítico (aumenta afrequência
cardíaca)
____
_
_
_
_
_
_;
Pancurônio ,._
�-
�6
-
-
-
-
-
-
-
Rocurônio 1-
/--'-
:
-
3-
-
�
Écomum adormuscularpós-cirúr
gica; podeocorrerhiperpotassemiae
aumentodas pressõesintraoculare
intragástrica. O fármacopode
desencadearhipertermia maligna.
O iníciorápidodaaçãotornaa
succinilcolinaútilparaaentubação
traqueal em pacientescomconteúdo
gástrico.
S . ., ,. l.h1
ucc1n11co1na @]
Tubocurarina ,.__;8--�
Vecurônio ,._
�-
�
-
-
-..
Figura 5.11
Mecanismo de ação dos fármacos blo
queadores neuromusculares despolari
zantes.
muscular e a duração de efeito pretendida. O início e a duração de
ação, bem como outras características dos fármacos bloqueadores
neuromusculares são mostrados na Figura 5.1 1 .
5. Efeitos adversos. Em geral, os fármacos são seguros com efeitos
adversos mínimos. Os efeitos adversos dos fármacos bloqueadores
neuromusculares específicos são mostrados na Figura 5.1 1 .
6. Interações de fármacos
a. Inibidores de colinesterase. Fármacos como neostigmina, fi
sostigmina, piridostigmina e edrofônio podem reverter a ação
dos bloqueadores neuromusculares não despolarizantes, mas,
com dosagens elevadas, os inibidores de colinesterase podem
causar um bloqueio despolarizante como resultado da concen
tração elevada de ACh na placa motora. Se o bloqueador neu
romuscular entrou no canal iônico, os inibidores de colinestera
se são incapazes de reverter o bloqueio.
b. Anestésicos hidrocarbonetos halogenados. Fármacos como
o halotano potencializam o bloqueio neuromuscular por exerce
rem umaação estabilizadora na JNM. Esses fármacos sensibili
zam a JNM aos efeitos dos bloqueadores neuromusculares.
c. Antibióticos aminoglicosídeos. Fármacos como a gentamici
na e a tobramicina inibem a liberação de ACh dos nervos co
linérgicos por competição com os íons de cálcio. Eles atuam
sinergicamente com o pancurônio e outros bloqueadores com
petitivos, aumentando o bloqueio.
d. Bloqueadores de canais de cálcio. Esses fármacos podem
aumentaro bloqueio neuromusculardos bloqueadores competi
tivos, bem como os bloqueadores despolarizantes.
B. Fármacos despolarizantes
Os fármacos bloqueadores despolarizantes atuam por despolarização da
membrana plasmática dafibra muscular, similarà ação daACh. Entretan
to, estesfármacos são mais resistentes à degradação pela AChE e assim
despolarizam as fibras musculares de modo mais persistente. A succi
nilcolina é o único relaxante muscular depolarizante usado atualmente.
1. Mecanismo de ação. O bloqueador neuromuscular despolarizan
te succinilcolina liga-se ao receptor nicotínico e atua como a ACh
despolarizando a junção (Figura 5.12). Ao contrário da ACh, que é
destruída instantaneamente pela AChE, o fármaco despolarizante
persiste em concentração elevada na fenda sináptica, permanecen
do ligado ao receptor por um tempo maior e causando uma estimu
lação constante do receptor. (Nota: a duração da ação da succinil
colina depende da sua difusão da placa motora e da hidrólise pela
pseudocolinesterase plasmática.) O fármaco despolarizante inicial
mente causa a abertura do canal de sódio associado ao receptor
nicotínico, o que resulta na despolarização do receptor (Fase 1). Isso
leva a abalos contráteis transitórios do músculo (fasciculações). A
ligação persistente torna o receptor incapaz de transmitir impulsos
adicionais. Com o tempo, a despolarização contínua dá origem a
uma repolarização gradual quando o canal de sódio se fecha ou
é bloqueado. Isso causa a resistência à despolarização (Fase 11) e
paralisia flácida.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-76-320.jpg)



![70 Clark, Finkel, Rey &Whalen
Receptor
nicotínico
Suprarrenal
Epinefrina
liberada
na circulação
sanguínea
o
•
Receptor
adrenérgico
,
Receptor
nicotínico
Norepinefrina
Receptor
adrenérgico
Orgãos efetores
Figura 6.2
Locais de ação dos agonistas adrenér-
•
glCOS.
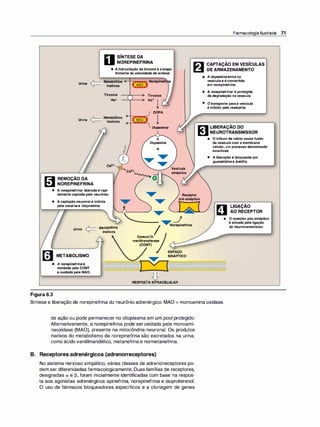
droxilase.
1
Essa é a etapa limitante da velocidade de síntese de
norepinefrina. A DOPA é descarboxilada pela dopadescarboxilase
(descarboxilase de L aminoácido aromático), formando dopamina
no citoplasma do neurônio pré-sináptico.
2. Armazenamento de norepinefrina em vesículas. A dopamina é en
tãotransportada parao interiordasvesículas porum sistematranspor
tador de aminas que também está envolvido na captação da norepi
nefrina pré-formada. Esse sistema é bloqueado pela reserpi
na (ver p.
96).A dopamina é hidroxilada paraformara norepinefri
na pela enzima
dopamina hidroxilase. (Nota: as vesículas sinápticas contêm dopami
na ou norepinefri
na mais trifosfato de adenosina [A
TP] e hidroxilase,
bem como outros cotransmissores.) Na suprarrenal, a norepinefri
na é
metilada à epinefrina e armazenada nas células cromafins junto com
norepinefrina. Sob estimulação, a suprarrenal libera cerca de 80°
/o de
epinefrina e 20°
/
o de norepinefrina diretamente na circulação.
3. Liberação de norepinefrina. A chegada do potencial de ação na
junção neuromuscular inicia a entrada de íons cálcio do líquido ex
tracelular para o axoplasma. O aumento no cálcio causa afusão das
vesículas com a membrana celular e a liberação de seu conteúdo
(exocitose) na sinapse. Fármacos como a guanetidi
na bloqueiam
essa liberação (ver p. 96).
4. Ligação aos receptores. A norepinefrina liberada das vesículas
sinápticas difunde-se através do espaço sináptico e se liga aos re
ceptores pós-sinápticos no órgão efetor ou aos receptores pré-si
nápticos no terminal nervoso. O reconhecimento da norepinefrina
pelos receptores inicia uma cascata de eventos no interior da célu
la, resultando na formação do segundo mensageiro intracelular que
atua como intermediário (transdutor) na comunicação entre o neuro
transmissore aação gerada no interiorda célula efetora. Receptores
adrenérgicos usam o monofosfato cíclico de adenosina (AMP0) como
segundo mensageiro
2
e o ciclo do fosfatidilinositol
3
para transduzir o
sinal em um efeito. A norepinefrina também se liga a receptores pré
-sinápticos que modulam a liberação do neurotransmissor.
5. Remoção da norepinefrina. A norepinefri
na pode 1) difundir-se
para fora da fenda sináptica e entrar na circulação geral; 2) ser me
tabolizada a derivados 0-metilados pela catecol-0-metiltransferase
associada à membrana pós-sináptica na fenda sináptica ou 3) ser
capturada por um sistema de transporte que a bombeia de volta
para o neurônio. A captação neuronal envolve uma ATPase ativada
por Na+ ou K+ que pode ser inibida pelos antidepressivos tricíclicos,
como imipramina, ou pela cocaína (ver Figura 6.3). O mecanismo de
captação da norepinefrina para o interior do neurônio pré-sináptico
é o mecanismo primário paraterminar com os seus efeitos.
6. Possíveis destinos da norepinefrina captada. Logo que a nore
pinefrina entra no citoplasma do neurônio adrenérgico, ela é capta
da para o interior da vesícula através do sistema transportador de
aminas e é sequestrada para futura liberação por outro potencial
1
Ver Capítulo 21 de Bioquímica /lustrada, 4ª edição, Artmed Editora, para
uma discussão sobre a síntese de DOPA.
2
Ver Capítulo 8 de Bioquímica /lustrada, 4ª edição, Artmed Editora, para uma
discussão sobre sistema de segundo mensageiro AMPc.
3Ver Capítulo 17 de Bioquímica /lustrada, 4ª edição, Artmed Editora, para
uma discussão sobre ciclo do fosfatidilinositol.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-80-320.jpg)
![72 Clark, Finkel, Rey &Whalen
rJ Adrenorreceptores a
Norepinefrina
Epinefrina lsoproterenol
• • •
' ' l
IDlID �
= �
=�
=
Receptoro:
Alta � Baixa
afinidade
afinidade
l]J Adrenorreceptores J
Epinefrina
lsoproterenol Norepinefrina
• • •
' ' l
IDfill=+ n
1
1--+ {
filfilj� =
Receptor�
Alta +- Baixa
afinidade
afinidade
Figura 6.4
Tipos de receptores adrenérgicos.
revelaram as identidades moleculares de inúmeros subtipos de recepto
res. Essas proteínas pertencem a uma família multigênica. Alterações na
estrutura primária dos receptores influenciam sua afinidade para vários
fármacos.
1 . Receptores a1 e a2• Os adrenorreceptores apresentam respostas
fracas ao agonista sintético isoproterenol, mas eles respondem às
catecolaminas naturais epinefri
na e norepinefrina (Figura 6.4). Para
os receptores a, a ordem de potência é epinefrina > norepinefri
na >> isoproterenol. Os adrenorreceptores a são subdivididos em
dois grupos, a1 e a2, com base nas suas afinidades por agonistas e
bloqueadores a. Por exemplo, os receptores a1 têm maior afinidade
por fenilefri
na do que os receptores a2• Ao contrário, a clonidina se
liga seletivamente aos receptores a2 e tem menor efeito nos recep
tores a1•
a. Receptores a1• Esses receptores estão presentes na membrana
pós-sinápticadosórgãos efetores e intermedeiam vários dos efei
tos clássicos-originalmente designados como a-adrenérgicos-,
envolvendo contração de músculo liso.A ativação dos receptores
a1 inicia uma série de reações por meio da fosfolipase C ativada
pela proteína G, resultando na formação de inositol1 ,4,5trifosfato
(IP3) e de diacilglicerol (DAG) do fosfatidilinositol. O IP3 inicia a
liberação de Ca
2
+ do retículo endoplasmático para o citosol, e o
DAG ativa outras proteínas no interiorda célula (Figura 6.5).
b. Receptores a2• Esses receptores, localizados primariamente
nas terminações nervosas pré-sinápticas e em outras células,
como a célula do pâncreas e certas células musculares lisas
vasculares, controlam respectivamente a liberação do neurome
diador adrenérgico e da insulina. Quando um nervo simpático
adrenérgico é estimulado, a norepinefrina liberada atravessa a
fenda sináptica e interage com o receptor a1• Uma porção da
norepinefrina liberada reage com os receptores a2 na membra
na neuronal (ver Figura 6.5). A estimulação desse receptor a2
promove retroalimentação inibitória da liberação de norepine
frina do neurônio adrenérgico estimulado. Esse efeito inibitório
diminui a liberação adicional do neurônio adrenérgico e serve
como um mecanismo modulador local para reduzir o débito de
neuromediador simpático quando há atividade simpática eleva
da. (Nota: neste caso, esses receptores atuam como autorre
ceptores inibitórios). Receptores a2 também são encontrados na
pré-sinapse dos neurônios parassimpáticos. A norepinefrina li
berada de um neurônio pré-sináptico simpático pode difundir-se
e interagir com esses receptores, inibindo a liberação de acetil
colina. (Nota: neste caso, tais receptores atuam como heterorre
ceptores inibitórios). Este é outro mecanismo modular local para
controlar a atividade autônoma em certas áreas. Em contraste
com os receptores a1, os efeitos da ligação com os receptores
a2 são mediados pela inibição da adenililciclase e pela redução
nos níveis intracelulares de AMPc.
c. Subdivisões adicionais. Os receptores a1 e a2 são subdividi
dos em a1A, a18, a1c e a10 e em a2A, a28, a2c e a20• Essa clas
sificação estendida é necessária para entender a seletividade
de alguns fármacos. Porexemplo, tansu/osina é um antagonista
seletivo a1A e é usada para o tratamento da hiperplasia benigna
de próstata. O fármaco é útil clinicamente, pois atua sobre os
receptores a1Aencontrados primariamente notrato urinário e na
próstata.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-82-320.jpg)







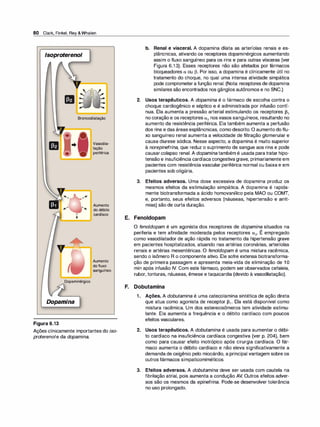


![C. Cocaína
A cocaína é o único anestésico local que bloqueia a ATPase ativada por
Na+/K+ (necessária para a captação neuronal da norepinefrina) na mem
brana celular dos neurônios adrenérgicos. Consequentemente, a norepi
nefrina acumula-se na fenda sináptica, resultando em aumento da ativi
dade simpática e potenciação das ações da epinefri
na e norepinefrina.
Por isso, pequenas doses de catecolaminas produzem efeitos bastante
intensificados em indivíduos que usam cocaínacomparados com os efei
tos naqueles que não usam.Além disso, a duração de ação da epinefrina
e da norepinefrina é prolongada. Semelhante às anfetaminas, a cocaína
pode aumentar a pressão arterial por ação agonista a1 e efeitos estimu
lantes �· (Nota: a cocaína como estimulante do SNC e como droga é
discutida nas pp. 126-127).
VI. AGONISTAS ADRENÉRGICOS DE AÇÃO MISTA
Agonistas adrenérgicos de ação mista induzem a liberação de norepinefrina
dos terminais pré-sinápticos, ativando também os receptores adrenérgicos na
membrana pós-sináptica (ver Figura 6.8).
A. Efedrina e pseudoefedrina
A efedri
na e a pseudoefedrina são alcaloides vegetais, hoje produzidos
sinteticamente. Ambas são adrenérgicas de ação mista. Elas não só li
beram a norepinefrina armazenada nos terminais nervosos (Figura 6.8),
como também estimulam diretamente os receptores a e �. Dessa for
ma, elas produzem vários efeitos adrenérgicos que são similares aos da
epinefrina, embora menos potentes. A efedrina e a pseudoefedri
na não
são catecóis e são maus substratos para MAO e COMT. Portanto, esses
fármacos têm ação longa. A efedrina e a pseudoefedrina têm excelente
absorção porvia oral e penetram no SNC; contudo, apseudoefedri
natem
efeitos menores no SNC. A efedrina é eliminada praticamente inaltera
da na urina, e a pseudoefedrina sofre biotransformação hepática incom
pleta antes de sua eliminação na urina. A efedrina aumenta a pressão
arterial sistólica e diastólica por vasoconstrição e estimulação cardíaca.
Ela produz broncodilatação, mas é menos potente do que a epinefri
na e
o isoproterenol nesse aspecto e produz seu efeito mais lentamente. No
passado, ela foi usada contra a asma para prevenir ataques (em vez de
tratar os ataques agudos), embora a maioria dos especialistas recomen
de outras medicações (ver Cap. 27). Ela produz uma leve estimulação do
SNC, aumentando o estado de alerta, diminuindo a fadiga e dificultando
o sono. Ela também melhora o desempenho atlético. A pseudoefedrina
é usada primariamente no tratamento da congestão nasal e sinusal e
congestão das trompas de Eustáquio, porvia oral. (Nota: o uso clínico da
efedrina está em declínio devido à disponibilidade de fármacos mais po
tentes e melhores que causam menos efeitos adversos. Os fitoterápicos
contendo efedrina [principalmente produtos contendo Ephedra] foram ba
nidos pelo FDA [Food and Drug Administration] nos EUA em abril de 2004
devido a reações cardiovasculares ameaçadoras à vida.) A pseudoefedri
naé convertida em metanfetamina ilegalmente. Por isso, os produtos que
contêm pseudoefedrina têm certas restrições, e a comercialização deve
ser mantida sob controle rígido nos EUA. Características importantes dos
agonistas adrenérgicos estão resumidas nas Figuras 6.15, 6.16 e 6.17.
Farmacologia Ilustrada 83
v==�
I
I
1
1
1
�
/
/
v
=
=;=
o --.
o
Figura 6.15
Arritmia
Cefaleia
Hiperatividade
Insônia
Náusea
Tremores
Alguns efeitos adversos observados
com os agonistas adrenérgicos.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-93-320.jpg)
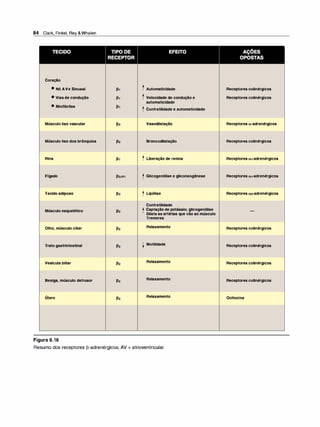


![, •
rener ICOS
1. RESUMO
Os antagonistas adrenérgicos (também denominados bloqueadores adrenér
gicos ou simpaticolíticos) ligam-se aos adrenorreceptores, mas não iniciam
os usuais efeitos intracelulares mediados pelos receptores. Esses fármacos
ligam-se reversível ou irreversivelmente ao receptor, evitando, assim, sua ati
vação pelas catecolaminas endógenas. Como os agonistas, os antagonistas
adrenérgicos são classificados de acordo com suas afinidades relativas para
os receptores O'. ou � no sistema nervoso periférico. Estes fármacos interferem
com as funções do sistema nervoso simpático. Numerosos antagonistas adre
nérgicos têm papeis importantes na clínica, primariamente para tratardoenças
associadas com o sistema cardiovascular. (Nota: antagonistas que bloqueiam
os receptores dopaminérgicos são mais importantes no SNC e, portanto, são
considerados na respectiva seção [ver p. 161].) Os fármacos bloqueadores
adrenérgicos discutidos neste capítulo estão resumidos na Figura 7.1 .
li. BLOQUEADORES o.-ADRENÉRGICOS
Fármacos que bloqueiam os adrenorreceptores O'. afetam profundamente
a pressão arterial. Como o controle simpático normal dos vasos ocorre em
grande parte por ações agonistas nos receptores O'.-adrenérgicos, o bloqueio
desses receptores reduz o tônus simpático dos vasos sanguíneos, resultando
em menor resistência vascular periférica. Isso induz a taquicardia reflexa re
sultante da redução da pressão arterial. A intensidade da resposta depende
do tonus simpático do indivíduo quando o fármaco é administrado. Os efei
tos são mais intensos no indivíduo em estação e menos naqueles que estão
deitados. Pacientes hipovolêmicos também tem respostas mais acentuadas.
(Nota: os receptores �. incluindo os adrenorreceptores �1 cardíacos, não são
afetados pelo bloqueio O'..) Os bloqueadores Q'.-adrenérgicos, fenoxibenzamina
a-BLOQUEADORES
Alfuzoslna
Doxazoslna
Fenoxlbenzamlna
Fentolamlna
Prazos/na
Tansuloslna
Terazoslna
lolmblna
P-BLOQUEADORES
Acebutolol
Atenolol
Betaxolol
Bisopro/oi
Carteolol
Carvedllol
Esmolo/
Labetalol
Metoprolol
Nado/oi
Neblvolol
Penbutolol
Plndolol
Propranolol
Ti
mo/oi
FÁRMACOS QUEAFETAM A CAPTAÇÃO OU
A LIBERAÇÃO DENEUROTRANSMISSORES
Guanetldlna
Reserplna
e fentolamina, têm aplicações clínicas limitadas. figura 7.1
Resumo dos fármacos bloqueadores,
bem como os que afetam a captação
ou a liberação do neurotransmissor.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-97-320.jpg)





![4. Efeitos adversos
a. Broncoconstrição. O propranolol apresenta um efeito adverso
grave e potencialmente fatal quando administrado a pacientes
com asma (Figura 7.9). A contração imediata da musculatura lisa
bronquiolar impede a entrada do ar nos pulmões. Mortes poras
fixiaforam registradas em asmáticos que inadvertidamente rece
beram o fármaco. Por isso, o propranolol nunca deve ser usado
no tratamento de qualquer indivíduo com DPOC ou asma.
b. Arritmias. O tratamento com J3-bloqueadores nunca deve ser
interrompido abruptamente devido ao risco de precipitar ar
ritmias cardíacas, que podem ser graves. Os J3-bloqueadores
devem ser retirados gradualmente ao longo de algumas sema
nas. O tratamento de longa duração com o J3-antagonista leva à
supersensibilização do receptor J3. Ao suspender o tratamento,
esse número aumentado de receptores pode agravar a angina
e/ou a hipertensão.
e. Comprometimento sexual. Como a função sexual masculina
ocorre pela ativação a-adrenérgica, os J3-bloqueadores não
afetam a ejaculação normal nem a função do esfíncter vesical
interno. Por outro lado, alguns homens reclamam de compro
metimento da atividade sexual. As razões não estão claras, e o
efeito pode ser independente do bloqueio do receptor J3.
d. Distúrbios no metabolismo. O bloqueio J3 diminui a glicogenó
lise e a secreção de glucagônio. Pode ocorrer hipoglicemia de
jejum.Além disso, os J3-bloqueadores podem prevenir os efeitos
contrarreguladores das catecolaminas durante a hipoglicemia.
A percepção de sintomas, como tremores, taquicardia e nervo
sismo, são embotados. (Nota: J3-bloqueadores cardiosseletivos
são preferidos no tratamento de asmáticos que usam insulina
[ver antagonistas 131 seletivos].) A principal função dos recepto
res J3 é mobilizar moléculas energéticas como os aminoácidos.
(Nota: as lipases nas células adiposas são ativadas, levando
ao metabolismo de triglicerídeos em ácidos graxos livres.) Os
pacientes que recebem J3-bloqueadores não seletivostêm maior
quantidade de lipoproteína de baixa densidade ("mau" coleste
rol), aumento de triglicerídeos e redução de lipoproteína de alta
densidade ("bom" colesterol). Por outro lado, o perfil lipídico sé
rico em pacientes com dislipidemia melhora com o uso de anta
gonistas 131 seletivos, como o metoprolol.
e. Efeitos no SNC. O propranolol provoca numerosos efeitos no
SNC, incluindo depressão, tonturas, letargia, fadiga, fraqueza, dis
túrbios visuais, alucinações, perda de memória de curta duração,
fragilidade emocional, sonhos intensos (incluindo pesadelos), di
minuição da performance e depressão manifestada por insônia.
f. Interações. Os fármacos que interferem ou inibem a biotransfor
mação do propranolo/, como cimetidina, f/uoxetina, paroxetina e ri
tonavir, podem potencializarseus efeitosanti-hipertensivos. Osfár
macos que estimulam ou induzem a sua biotransformação, como
barbitúricos, fenitoína e rifampicina, podem reduzirseus efeitos.
B. Timolol e nadolol: antagonistas J3 não seletivos
O timo/oie o nado/o/também bloqueiam adrenorreceptores J3, e 132 e são
mais potentes do que o propranolol. O nado/oitem uma duração de ação
muito longa (ver Figura 7.7). O timo/oi reduz a produção de humor aquo-
Farmacologia Ilustrada 93
Fadiga
,,
"
Broncoconstrição ......
'-
...
;r
,,
Disfunção sexual
Arritmias
(após interrupção
abrupta do uso)
Figura 7.9
I
Je'
.-
...
.....
Efeitos adversos comumente observa
dos em indivíduos tratados com propra
nolol.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-103-320.jpg)
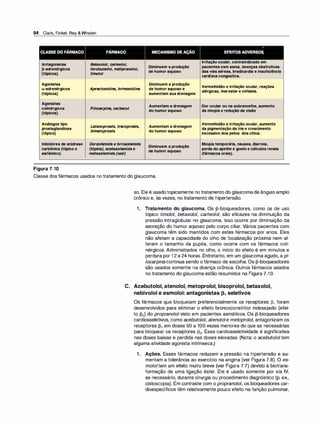
















![112 Clark, Finkel, Rey & Whalen
TRATAMENTO DA DEPENDêNCIA DO ÁLCOOL
Acsmprosato
Oissuff
iram
Na/trexona
Figura 9.1 (Continuação)
Resumo dos fármacos ansiolíticos e hip
nóticos.
As benzodiazepinas são relativa
mente seguras, pois a dose letal é
mais de 1.000vezes maior do que
a dose terapêutica típica.
Morfi
na
Clorpromazi
na
Fenobarbital
Oiazepam
Figura 9.2
-
�
�
�
�
�
�
...... ,...,__,
o 20 40 1.000
Relação = dose letal
dose eficaz
Relação entre a dose letal e a dose
eficaz para morfina (um opioide, ver
Capítulo 14), c/orpromazina (um neuro
léptico, ver Capítulo 13) e os fármacos
ansiolíticos e hipnóticos, fenobarbital e
diazepam.
na interface da subunidade a. e da subunidade 'Y2 (ver Figura 9.3). (Nota:
os locais de ligação, algumas vezes, são denominados receptores ben
zodiazepínicos. Dois subtipos de receptores benzodiazepínicos comu
mente encontrados no SNC são designados como receptores BZ1 e BZ2,
dependendo se na sua estrutura se encontram subunidades a.1 ou a.2,
respectivamente.) A localização dos receptores BZ no SNC é comparável
à dos neurônios GABA. A ligação do GABA ao seu receptor abre o canal
de c1-, o que aumenta a condutância do íon (ver Figura 9.3). Os benzo
diazepínicos aumentam a frequência da abertura dos canais produzida
pelo GABA. O influxo de c1- causa uma leve hiperpolarização que afasta
o potencial pós-sináptico do valor limiar e, assim, inibe a formação de
potenciais de ação. (Nota: a ligação do benzodiazepínico ao seu receptor
aumentará a afinidade do GABA por seus locais de ligação [e vice-versa]
sem realmente alterar o número total de locais.) Os efeitos clínicos dos
vários benzodiazepínicos se correlacionam bem com cada afinidade de
ligação do fármaco pelo complexo receptor GABA-canal de íon cloreto.
B. Ações
Os benzodiazepínicos não têm atividade antipsicótica nem ação analgé
sica e não afetam o SNA. Todos os benzodiazepínicos apresentam ações
em maior ou menor intensidade citadas a seguir.
1. Redução da ansiedade. Em doses baixas, os benzodiazepínicos
são ansiolíticos. O efeito é atribuído à potenciação seletiva datrans
missão GABAérgica em neurônios que têm a subunidade a.2 no re
ceptor GABAA, inibindo, assim, os circuitos neuronais no sistema
límbico cerebral.
2. Ações hipnóticas e sedativas. Todos os benzodiazepínicos usa
dos para tratar ansiedade têm alguma propriedade sedativa, e al
guns podem produzir hipnose (sono produzido "artificialmente") em
doses mais elevadas. Seus efeitos são mediados pelos receptores
a.1-GABAA.
3. Amnésia anterógrada. O bloqueio temporário da memória com o
uso dos benzodiazepínicos também é mediado pelos receptores a.1_
-GABAA e diminui a capacidade do paciente de aprender e de for
mar novas memórias.
4. Anticonvulsivantes. Vários dos benzodiazepínicos têm atividade
anticonvulsivante e alguns são usados para tratar epilepsia (estado
epiléptico), e outros, distúrbios convulsivos. Esse efeito é parcial
mente mediado pelos receptores a.1-GABAA.
5. Relaxamento muscular. Em doses elevadas, os benzodiazepínicos
diminuem a espasticidade do músculo esquelético, provavelmente
aumentando a inibição pré-sináptica na medula espinal, onde predo
minam os receptores a.2-GABAA. O baclofeno é um relaxante muscu
lar que parece atuar nos receptores GABA8 na medula espinal.
C. Usos terapêuticos
Os benzodiazepínicos individuais mostram pequenas diferenças em suas
propriedades ansiolíticas, anticonvulsivantes e sedativas. Contudo, a du
ração de açãovaria bastante no grupo, e considerações farmacocinéticas
são importantes na escolha de uma delas.
1. Ansiedade. Os benzodiazepínicos são eficazes no tratamento dos
sintomas da ansiedade secundária ao transtorno de pânico, do
transtorno de ansiedade generalizada (TAG), do transtorno de an-](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-122-320.jpg)
![rJ Receptor vazio
(sem agonistas)
l]J Receptor ligado
com GABA
GABA
+
+
_ _ ...-.
............_
,
)
r
+
+
Farmacologia Ilustrada 1 1 3
O receptor vazio é inativo,
e o canal de cloreto acoplado
está fechado.
A ligação do GABA causa
abertura do canal de cloreto,
levando à hiperpolarização
da célula.
B Receptor
ligado com GABA
e benzodiazepínico
GABA
Benzodiazepínico
A entrada de c1- hiperpolariza a célula, tornando
mais difícil sua despolarização e, por isso, reduz
a excitabilidade neuronal.
Figura 9.3
+ + + + + +
l�
-"-..A..A.A-/
- -
Diagrama esquemático do complexo canal íon cloreto-GABA-benzodiazepínico.
siedade social e por performance, do transtorno de estresse pós
-traumático, dotranstorno obsessivo-compulsivo e da ansiedade ex
trema encontrada, às vezes, com fobias específicas, como o medo
de voar. Os benzodiazepínicos também são úteis no tratamento da
ansiedade que acompanha algumas formas de depressão e es
quizofrenia. Esses fármacos não devem ser usados para aliviar o
estresse normal da vida diária. Eles devem ser reservados para a
ansiedade grave e contínua e, então, usados somente em períodos
curtos devido ao seu potencial de vício. Os benzodiazepínicos de
ação mais longa, como clonazepam, /orazepam e diazepam, são
preferidos nos pacientes com ansiedade que exigem tratamento por
tempo prolongado. Os efeitos ansiolíticos dos benzodiazepínicos
são menos sujeitos à tolerância do que os efeitos sedativo e hipnó
tico. (Nota:tolerância- isto é, diminuição da respostacom doses re
petidas - ocorre quando o uso se estende por mais do que uma ou
duas semanas. Hátolerânciacruzada entre esse grupo defármacos
e o etanol. Foi demonstrado que a tolerância está associada a uma
diminuição na densidade de receptores GABA.) Para o transtorno
de pânico, o alprazolam é eficaz para tratamentos curtos ou longos,
embora possa causar reações de abstinência em cerca de 30°
/
o dos
pacientes.
2. Distúrbios musculares. O diazepam é útil no tratamento de espas
mos dos músculos esqueléticos, como os que ocorrem no estira
mento, e no tratamento daespasticidade devidaa doenças degene
rativas, como esclerose múltipla e paralisia cerebral.
A ligação do GABA é
potenciada pelo
benzodiazepínico, resultando
em maior entrada de
íons cloreto.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-123-320.jpg)


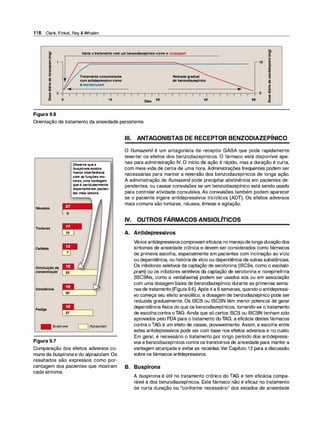




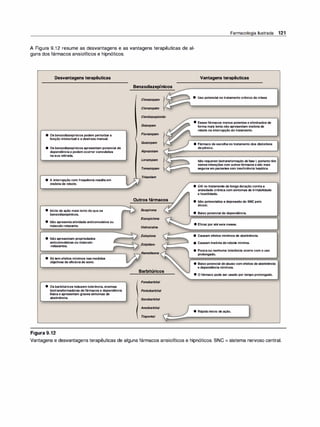





![tes, a interação com canais de potássio pode contribuir com a pro
priedade da cocaína de causararritmias cardíacas. (Nota: a cocaína
é o único anestésico local que causa vasoconstrição. Esse efeito é
responsável pela necrose e perfuração do septo nasal observadas
com a inalação crônica de pó de cocaína.)
4. Farmacocinética. Com frequência a cocaína é autoadministrada
por via oral (mascada), por inalação, fumo ou por injeção IV. O pico
ocorre em 15 a 20 minutos após a inalação do pó de cocaína, e o
efeito desaparece em 1 a 1,5 hora. Efeitos rápidos, mas de curta
duração, são obtidos por meio de injeção IV ou por fumar a forma
base livre da droga (crack). Como o início do efeito é mais rápido, o
potencial de dose excessiva e dependência é maior com a injeção
IV e com o fumo do crack. A cocaína é rapidamente desesterificada
e desmetilada à benzoilecgonina, que é excretada na urina. A de
tecção dessa substância na urina identifica o usuário.
5. Efeitos adversos
a. Ansiedade. A resposta tóxica à ingestão aguda de cocaína
pode precipitar reações de ansiedade que incluem hipertensão,
taquicardia, sudoração e paranoia. (Nota: ocorre pouca tolerân
cia aos efeitos tóxicos da cocaína no SNC, [p. ex., convulsões]
mesmo com uso prolongado). Devido à irritabilidade, vários
usuários usam a cocaína com álcool. Um produto da biotrans
formação da cocaína e do etanol é o cocaetileno, que também é
psicoativo e, acredita-se, contribui para a cardiotoxicidade.
b. Depressão. Como com todos os fármacos estimulantes, a esti
mulação do SNC pela cocaína é seguida por um período de de
pressão mental. Os adietas abstêmios de cocaína apresentam
depressão física e emocional, bem como agitação. A agitação
pode sertratada com benzodiazepínicos ou fenotiazinas.
c. Tóxicos. A cocaína pode provocar convulsões, bem como ar
ritmias cardíacas fatais (Figura 10.8). O uso de diazepam IV
pode ser necessário para controlar as convulsões induzidas por
cocaína. A incidência de infarto do miocárdio em usuários de
cocaína não está relacionada com a dose, a duração do uso
ou a via de administração. Não há marcador para identificar os
indivíduos que podem ter efeitos cardíacos ameaçadores à vida
após o uso de cocaína.
E. Anfetamina
A anfetamina é uma amina simpática que apresenta efeitos neurológi
cos e clínicos similares aos da cocaína. A dextroanfetamina é o principal
membro dessa classe de compostos. A metanfetamina (também conheci
da como 'speed'nos EUA) é um derivado da anfetamina disponível para
uso sob prescrição. Ela pode ser fumada e é preferida por vários adie
tas. A 3,4-metileno-dioxi-metanfetamina (também denominada MDMA
ou ecstasy), um derivado sintético da metanfetamina com propriedades
alucinogênicas e estimulantes, é discutida na p. 567.
1 . Mecanismo de ação. Como a cocaína, os efeitos da anfetamina
no SNC e no sistema nervoso periférico são indiretos, isto é, am
bos dependem da elevação dos níveis de catecolaminas nasfendas
sinápticas. A anfetamina, contudo, obtém seu efeito liberando esto
ques intracelulares de catecolaminas (Figura 10.9). Como a anfeta
mina também inibe a MAO, níveis elevados de catecolaminas são
V
Figura 10.8
Farmacologia Ilustrada 127
Euforia
Taquicardia
1 I
' /"
-
--
-
Agitação
Hipertensão
Dispneia
Convulsão
Arritmias
cardíacas
Insuficiência
respiratória
Morte
Principais efeitos do uso da cocaína.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-137-320.jpg)
![128 Clark, Finkel, Rey & Whalen
m Sem anfetamina
RESPOSTA
l]J Com anfetamina
Figura 10.9
RESPOSTA
AUMENTADA
Mecanismo de ação da anfetamina.
facilmente liberados para as fendas sinápticas. Apesar da diferença
no mecanismo de ação, os efeitos comportamentais da anfetamina
e de seus derivados são similares aos da cocaína.
2. Ações
a. SNC. Os principais efeitos comportamentais da anfetamina re
sultam da liberação de dopamina e de norepinefrina. A anfeta
mina estimula todo eixo cerebrospinal, córtex, tronco cerebral
e bulbo. Isso aumenta o estado de alerta, diminui a fadiga e
o apetite e causa insônia. Esses efeitos da anfetamina e seus
derivados justificam seu uso no tratamento de crianças hipe
rativas, para a narcolepsia e o controle do apetite. Em doses
elevadas, a anfetamina pode causar psicoses e convulsões.
b. Sistema nervoso simpático. Além da sua ação acentuada no
SNC, a anfetaminaatua no sistema adrenérgico estimulando in
diretamente os receptores com a liberação de norepinefrina.
3. Usos terapêuticos. Os fatores que limitam a utilidade terapêutica
da anfetamina incluem a dependência psico e fisiológica similar à
da cocaína e, com o uso crônico, o desenvolvimento de tolerância
aos efeitos eufóricos e anorexígenos.
a. Distúrbio de hiperatividade com déficit de atenção (DHDA).
Algumas crianças jovens são hipercinésicas e não têm capaci
dade de se concentrar em uma atividade por mais que poucos
minutos.A dextroanfetaminae o derivado da anfetamina, metilfe
nidato, são capazes de diminuirafalta de atenção e aliviarvários
dos problemas comportamentais associados a essa síndrome,
diminuindo também a hipercinesia que essas crianças apresen
tam. A lisdexanfetamina é um pró-fármaco que é convertido no
fármaco ativo dextroanfetamina depois da absorção no TGI. A
lisdexanfetamina prolonga a atenção, permitindo melhor adapta
ção à atmosfera escolar. A atomoxetina é um fármaco não esti
mulante aprovado para o DHDA em crianças e adultos. (Nota: a
atomoxetina não deve sertomada por indivíduos sob tratamento
com IMAO e nem por pacientes com glaucoma de ângulo estrei
to.) Ao contrário do metilfenidato, que bloqueia a captação de
dopamina, a atomoxetinaé um inibidorda captação de norepine
fri
na, por isso, não causa vício e não é um fármaco controlado.
b. Narcolepsia. A narcolepsiaé um distúrbio do sono relativamen
te raro caracterizado por incontroláveis surtos de sono durante
o dia. Algumas vezes, é acompanhado de catalepsia, perda do
controle muscular, ou mesmo paralisia provocadas por emo
ções fortes, como risadas. Contudo, é a sonolência do paciente
que é combatida com fármacos como anfetamina ou metilfeni
dato. Recentemente, novos fármacos, modafinila e seu deriva
do enantiômero-R, armodafinila, se tornaram disponíveis para
tratar a narcolepsia. A modafinila produz menos efeitos psico
ativos e eufóricos, bem como menos alterações do humor, da
percepção, do pensamento e das sensações, típicas em outros
estimulantes do SNC. Ela provoca vigília. O seu mecanismo de
ação permanece incerto, mas pode envolver os sistemas adre
nérgicos e dopaminérgicos, embora tenha sido demonstrado
ser diferente do mecanismo de ação da anfetamina. A moda
finila é eficaz por via oral. Ela distribui-se por todo o organismo
e sofre extensa biotransformação hepática. Os metabólitos são
excretados na urina. Cefaleia, náusea e rinite são os efeitos ad-](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-138-320.jpg)


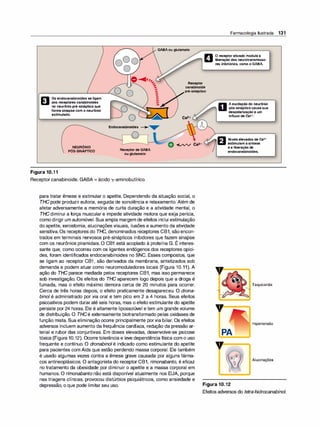





![A. Características comuns dos anestésicos inalatórios
Os anestésicos inalatórios modernos são fármacos não inflamáveis e não
explosivos que incluem o óxido nitroso, bem como um grande número de
hidrocarbonetos halogenados voláteis. Como grupo, esses fármacos dimi
nuem a resistência cerebrovascular, resultando em aumento da perfusão
do cérebro. Eles também causam broncodilatação e diminuem o volume
-minuto (volume de ar que se move para dentro ou para fora dos pulmões
por unidade de tempo) e a vasoconstrição pulmonar hipóxica (aumento da
resistênciavascularpulmonarem regiões malventiladas dos pulmões, o que
permite o direcionamento do fluxo sanguíneo pulmonar para regiões com
maior teor de oxigênio). O movimento desses fármacos desde os pulmões
até os diferentescompartimentos corporais depende de sua solubilidade no
sangue e nos tecidos, bem como no fluxo sanguíneo. Esses fatores desem
penham importante papel não só na indução, mastambém na recuperação.
B. Potência
A potência dos anestésicos inalatórios édefinidaquantitativamente como a
concentraçãoalveolar mínima (CAM). Essa é a concentração do gás anes
tésico no ar expirado, necessária para eliminar o movimento em 50°
/
o dos
pacientes submetidos a uma incisão cutânea padronizada. (Nota: a CAM
é a dose eficaz média [DE50] do anestésico.) A CAM normalmente é ex
pressa como a porcentagem de gás na mistura necessária para alcançar
o efeito. Numericamente, a CAM é pequena para os anestésicos potentes,
como o sevoflurano, e grande para os menos potentes, como o óxido nitro
so (N02). Assim, o inverso da CAM é um índice de potência do anestésico.
Osvalores da CAM são úteis na comparação de efeitos farmacológicos de
diferentesanestésicos, porque um CAM alto é igual à baixa potência (Figu
ra 1 1 .4). Note que só o óxido nitroso não consegue produzir umaanestesia
completa, pois a mistura com oxigênio suficiente não consegue alcançar
seu valor CAM. Quanto mais lipossolúvel é o anestésico, menor é a con
centração necessária para produzir anestesia e, assim, maior é a potência
do anestésico. Fatores que podem aumentar a CAM (e tornar o paciente
menos sensível) incluem hipertermia (acima de 42°C), fármacos que au
mentam as catecolaminas no SNC e abuso crônico de etanol. Fatores que
podem reduzir a CAM (e tornar o paciente mais sensível) incluem aumen
to da idade, hipotermia, gestação, sepse, intoxicação aguda com etanol,
administração concomitante de anestésicos IV e agonistas de receptores
a2-adrenérgicos (como a clonidina e a dexmedetomidi
na).
C. Absorção e distribuição dos anestésicos inalatórios
O principal objetivo da anestesia por inalação é obter uma pressão parcial
cerebral (Pcr) constante e ótima do anestésico inalado (pressão parcial
de equilíbrio entre os alvéolos [PA] e o cérebro [Pcrl). Assim, os alvéolos
são "ajanela para o cérebro"dos anestésicos inalados. A pressão parcial
de um gás anestésico na origem da via respiratória é a força que leva o
anestésico para o interiordo espaço alveolar e dali para o sangue, o qual
transporta o fármaco para o cérebro e paravários outros compartimentos
do organismo. Como os gases se movem de um compartimento para o
outro de acordo com os gradientes de pressão parcial, o equilíbrio é al
cançado quando a pressão parcial em cada um desses compartimentos
é equivalente àquela da mistura inalada. (Nota: no equilíbrio, a pressão
parcial alveolar= pressão parcial arterial = pressão parcial cerebral, ou PA
= Pª = Per·) O tempo necessário para se alcançar esse estado de equilí
brio é determinado pelos seguintes fatores:
1. Saturação alveolar (wash-in). Esse termo se refere à substituição
dos gases pulmonares normais pela mistura anestésica inalada. O
tempo necessário para esse processo é diretamente proporcional
Farmacologia Ilustrada 137
Halotano
Sevoflurano 2º/o
Desflurano
Óxido
nitroso
o 2 4
CAM
6
1 t
'
100
Porcentagemdegásanestésico
Figura 11.4
Concentrações alveolares mínimas
(CAM) dos gases anestésicos.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-147-320.jpg)

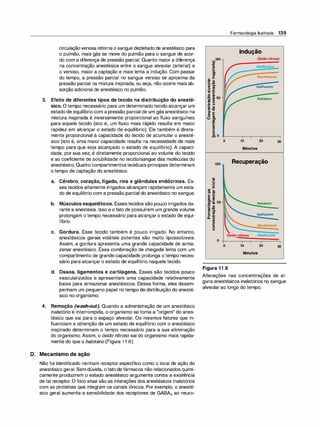
![140 Clark, Finkel, Rey & Whalen
a Sem anestésico
o
GABA
+ +
Aligação do GABAcausa
abertura do canal de íons
cloreto, levando à
hiperpolarização da célula.
.-1 + +
/
' 1
y· - -
c1+
ft Na presença de anestésico
lill inalatório
Aligação do GABAé
aumentada pelo anestésico
inalatório, resultando em
maior entrada de íons cloreto.
�
�-
GABA
+ + + + + +
Aentrada de CI- hiperpolariza
a célula, tornando mais difícil
a despolarização e, assim,
reduzindo a excitabilidade neural.
Figura 1 1 .7
Exemplo de modulação em um canal
de membrana estimulado por ligante
modulado por anestésicos inalatórios.
GABA = ácido -y-aminobutírico; CI- =
íons cloreto.
transmissor GABA em concentrações clinicamente eficazes do fármaco.
Isso causa um prolongamento da corrente inibitória de íons cloreto após
um pulso de liberação de GABA, diminuindo, assim, a excitabilidade neu
ronal pós-sináptica (Figura 1 1 .7). Outros receptores também são afetados
pelos anestésicos voláteis. Por exemplo, a atividade dos receptores inibi
tórios de glicina nos neurônios motores da medula espinal aumenta. Além
disso, os anestésicos inalatórios bloqueiam a corrente pós-sináptica exci
tatória dos receptores nicotínicos. O mecanismo pelo qual os anestésicos
realizam essas funções moduladoras ainda não foi explicado.
E. Halotano
Esse fármaco é o protótipo com o qual os novos anestésicos inalatórios
têm sido comparados. Quando o halotano foi introduzido, sua capacidade
de induzir rapidamente o estado anestésico e permitir rápida recuperação
-e o fato de não ser explosivo-tornaram-no o anestésico de primeira es
colha. No entanto, com o reconhecimento dos efeitos adversos discutidos
a seguir e a disponibilidade de outros anestésicos que causam menos
complicações, o halotanofoi amplamentesubstituído nos Estados Unidos.
1. Usos terapêuticos. O halotano é um anestésico potente, mas é um
analgésico relativamente fraco. Assim, normalmente ele é coadminis-
,
trado com óxido nitroso, opioides ou anestésicos locais. E um potente
broncodilatador. O halotano relaxa os músculos esqueléticos e uterino
e pode serutilizado em obstetríciaquando há indicaçãode relaxamen
to uterino. Ele não é hepatotóxico em pacientes pediátricos (apesar
do efeito potencial em adultos, vera seguir), fato que, combinado com
seu odor agradável, o torna uma boa escolha para indução inalatória
em crianças, embora o sevof/urano seja atualmente o fármaco de es
colha para a indução inalatória, se o custo nãofor um fator limitante.
2. Farmacocinética. O halotano é biotransformado por oxidação em
hidrocarbonetos tóxicos aos tecidos (por exemplo, trifluoretanol) e
íon brometo. Essas substâncias podem ser responsáveis pela rea
ção de toxicidade que alguns pacientes (especialmente mulheres)
desenvolvem após a anestesia com esse fármaco. Essa reação
inicia com febre, seguida de anorexia, náusea e êmese, e o pa
ciente pode demonstrar sinais de hepatite. (Nota: embora a incidên
cia dessa reação seja baixa - aproximadamente de 1 em 10.000
indivíduos -, 50°
/
o dos pacientes atingidos podem morrer por necro
se hepática. Para evitar essa condição, a anestesia com halotano
não é repetida em intervalos menores do que 2 a 3 semanas. Foi
descrito que todos os anestésicos inalatórios halogenados causam
hepatite, mas em uma incidência muito menordo que com halotano.
Por exemplo, o isof/uranotem uma incidência de 1 :500.000.)
3. Efeitos adversos
a. Efeitos cardíacos. Como outros hidrocarbonetos halogenados,
o halotano é vagomimético e causa bradicardia atropina-sen
sível. Além disso, ele apresenta a propriedade indesejável de
causar arritmias cardíacas. (Nota: isso é especialmente grave
quando se desenvolve hipercapnia [aumento na pressão par
cial arterial de dióxido de carbono] devido à diminuição da ven
tilação alveolar ou ao aumento da concentração plasmática de
catecolaminas.) O halotano, como outros anestésicos haloge
nados, produz hipotensão concentração-dependente. Se houver
necessidade de combater um episódio de hipotensão excessiva
durante a anestesia com halotano, é recomendado utilizar um
vasoconstritor de ação direta, como a fenilefrina.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-150-320.jpg)

















![tolamina ou a prazosina são úteis no tratamento da hipertensão induzida
portiramina. (Nota: o tratamento com IMAO pode ser perigoso em pacien
tes gravemente deprimidos e com tendência suicida. O consumo proposi
tal de alimentos contendo tiramina é uma possibilidade.) Outros possíveis
efeitos adversos do tratamento com IMAO incluem sonolência, hipotensão
ortostática, visão borrada, xerostomia, disúria e constipação. Os IMAOs e
os ISCSs não devem sercoadministrados devido ao risco de"síndrome de
serotonina" que ameaça a vida. Ambos os tipos de fármacos precisam do
período de eliminação, de duas semanas no mínimo, antes da administra
ção do fármaco do outro tipo, com exceção da fluoxetina, a qual deve ser
suspensa no mínimo seis semanas antes de iniciaro IMAO. A associação
de IMAOs com bupropiona pode causar convulsões. A Figura 12.1O resu
me os efeitos adversos dos fármacos antidepressivos.
VIII. TRATAMENTO DA MANIA E DO DISTÚRBIO BIPOLAR
O tratamento do transtorno bipolar aumentou em anos recentes, devido ao
maior reconhecimento do transtorno e também pelo aumento do número de
medicamentos disponíveis para o tratamento, aprovados pelo FDA.
A. Lítio
Os sais de lítio são usados profilaticamente para o tratamento de pacien
tes maníaco-depressivos e nos episódios de mania, e, por isso, são con
siderados como "estabilizadores do humor". O lítio é eficaz no tratamento
de 60 a 80°
/
o dos pacientes que exibem mania e hipomania. Emboravários
processos celulares sejam alterados pelos sais de lítio, o seu mecanismo
de ação segue desconhecido. (Nota: acredita-se que o lítio atenue os si
nais via receptores acoplados ao sistema desegundo mensageiro bifosfa
to de fosfatidilinositol [PIP2]. O lítio interfere com a ressíntese [reciclagem]
do PIP2, levando à sua depleção relativa na membrana neuronal do SNC.
Os níveis de PIP2 nas membranas periféricas não são afetados pelo lítio.)
Ele é administrado por via oral, e o íon é excretado pelos rins. Os sais de
lítio podem ser tóxicos. O fator de segurança e o índice terapêutico são
extremamente baixos comparáveis aos da digoxina. Os efeitos adversos
podem incluir cefaleia, xerostomia, polidipsia, polifagia, distúrbios gastrin
testinais (o lítio deve ser administrado com alimentação), tremor fino nas
mãos, tonturas, fadiga, reações dérmicas e sedação. Os efeitos adversos
devidos a níveis plasmáticos mais elevados incluem ataxia, fala enrola
da, tremores grosseiros, confusão e convulsões. Xerostomia, polidipsia e
poliúria são queixas frequentes. (Nota: o diabetes insípido que resulta do
uso do lítio pode ser tratado com amilorida.) A função tireoideana pode
diminuir e deve ser monitorada. O lítio não causa efeitos observáveis em
indivíduos normais. Ele não é um sedativo, euforizante ou depressor.
B. Outros fármacos
Vários fármacos antiepilépticos, incluindo de modo destacado carbama
zepina, ácido valproico e lamotrigina, foram identificados como "estabili
zadores do humor'', aprovados pelo FDA, e são usados com sucesso no
tratamento do transtorno bipolar. Outros fármacos que podem amenizar
os sintomas de mania incluem os antipsicóticos antigos (p. ex., clorpro
mazina e haloperido� e novos. Os antipsicóticos atípicos (risperidona,
olanzapina, zi
prasidona, aripiprazol, asenapina e quetiapina) também fo
ram aprovados pelo FDA para o controle da mania. Os benzodiazepínicos
são usados com frequência como auxiliares do tratamento para a esta
bilização aguda dos pacientes com mania. (Ver os respectivos capítulos
desses psicotrópicos para descrição mais detalhada.)
Farmacologia Ilustrada 159
Distúrbios
gastrintestinais
Sedação;podeser
útilcontraaagitação
INIBIDORESSELETIvos
DACAPTAÇÃO
DESEROTONINA
Citalopram -
Escitalopram
Fluoxetina
.
Fluvoxamina
Paroxetina -
-
Sertralina -
-
INIBIDORESDACAPTAÇÃO
DESEROTONINA
ENOREPINEFRINA
Duloxetina -
-
Venlafaxina -
Desven/afaxina -
-
ANTIDEPRESSIVOs
ATÍPICOS
Bupropiona
. .
Mirtazapina �
.
Nefazodona -
-
Trazodona
ANTIDEPRESSIVOs
TRIC[CLICOSIPOLIC[CLICOS
- Amitriptilina
-
Amoxapina
Clomipramina
Desipramina
-
Doxepina
- -
-
-
.
lmipramina
- -
.
Maprotilina
Nortriptili
na
Protriptilina
- Trimipramina
INIBIDORESDA
MONOAMINAOXIDASE
Fene/zina
Tranilcipromina
Ganho de
massa
Potencialelevado
parahipotensão
ortostática corporal
Figura 12.1O
Efeitos adversos de alguns fármacos
usados no tratamento da depressão.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-169-320.jpg)












![172 Clark, Finkel, Rey & Whalen
v===l
PA
Hipotensão
,,
Disforia
(ansiedade,
depressão e
mal-estar)
,,
Sedação
,,
Constipação
Retenção
urinária
Náusea
v=
V
Potencial
para
dependência
(vício)
Depressão
respiratória
Figura 14.5
�''
Z z
o
o
Efeitos adversos comumente observa
dos em indivíduostratados com opioides.
b.
e.
nas quais há dor e o sono é necessário, eles podem ser utili
zados para complementar a propriedade indutora de sono das
benzodiazepinas, como o temazepam. (Nota: normalmente os
sedativos-hipnóticos não são analgésicos e podem ter seu efei
to sedativo diminuído na presença de dor.)
Tratamento da diarreia. A morfina reduz a motilidade e au
menta o tônus da musculatura lisa circular intestinal. (Nota: isso
pode causar constipação. Produtos contendo morfina, como a
tintura de ópio e a tintura canforada de ópio [paregórico], têm
sido usados contra a diarreia.)
Alívio da tosse. Ainda que a morfina suprima o reflexo da tosse,
a codeína ou o dextrometorfano são mais utilizados com esse
objetivo. A codeína apresenta maior ação antitussígena do que a
morfina.
d. Tratamento do edema pulmonar agudo. A morfi
na por via IV
alivia a dispneia causada por edema pulmonar associado à fa
lência ventricular esquerda - possivelmente pelo seu efeito va
sodilatador.
4. Farmacocinética
a. Administração. Por causa da significativa biotransformação de
primeira passagem da morfina no fígado, as injeções IM, SC ou
IV produzem as respostas mais confiáveis. A absorção da mor
fina doTGI é lenta e errática. Quando a morfina é usada por via
oral, em geral é administrada em uma forma de liberação lenta
,
para obter níveis plasmáticos mais consistentes. E importante
notar que a morfi
na tem perfil farmacocinético linear. Este com
portamento farmacocinético permite que a dosagem seja mais
previsível e flexível. (Nota: nos casos de dor crônica associada
à doença neoplásica, é comum utilizar os comprimidos orais de
liberação lenta ou bombas que permitem ao paciente controlar
a dor pela autoadministração.)
b. Distribuição. A morfina entra rapidamente em todos os teci
dos do organismo, inclusive no feto, e não deve ser utilizada
para analgesia durante o trabalho de parto. Recém-nascidos
de mães adietas apresentam dependência física de opioides,
manifestando sintomas de abstinência se o opioide não for ad
ministrado. Somente uma pequena fração de morfina atravessa
a barreira hematencefálica, já que a morfinaé o fármaco menos
lipofílico dos opioides comuns. Nesse aspecto, a morfina con
trasta com os opioides mais lipossolúveis, como a fentanila, e a
metadona, que penetram rapidamente o cérebro.
e. Destino. A morfina é conjugada com ácido glicurônico no fí
gado. A morfina-6-glicuronato é um analgésico muito potente,
e o conjugado na posição 3 (morfina-3-glicuronato) não tem
atividade opioide, mas parece causar efeitos neuroexcitatórios
observados com doses altas de morfi
na. Os conjugados são
excretados primariamente na urina e pequenas quantidades
aparecem na bile. A duração de ação da morfina é de 4 a 6 ho
ras quando administrada sistemicamente em indivíduos nunca
expostos, e é consideravelmente mais longa quando injetada
por via epidural, porque sua baixa lipofilicidade retarda a redis
tribuição do espaço epidural. (Nota: a idade do paciente pode
influenciar a resposta à morfina. Pacientes idosos são mais sen
síveis ao efeito analgésico, possivelmente devido à diminuição](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-182-320.jpg)
![do metabolismo ou a outros fatores, como a diminuição da mas
sa corporal, da função renal, etc. Eles devem receber dosagens
menores. Os neonatos não devem receber morfinadevido à sua
baixa capacidade de conjugação.)
5. Efeitos adversos. Pode ocorrer grave depressão respiratória e re
sultar em óbito o envenenamento agudo com opioides. Um efeito
grave é a parada das trocas respiratórias em pacientes com enfise
ma ou corpulmonale. (Nota: se a morfina for utilizada nesses indi
víduos, a respiração deve ser cuidadosamente monitorada.) Outros
efeitos incluem êmese, disforia e efeitos hipotensivos acentuados
por histamina (Figura 14.5). A elevação da pressão intracraniana,
particularmente em lesões na cabeça, pode ser grave. A morfina
aumenta a isquemia cerebral e espinal. Na hiperplasia prostática
benigna, a morfina pode causar retenção urinária aguda. Pacien
tes com insuficiência suprarrenal ou mixedema podem apresentar
efeitos dos opioides mais intensos ou prolongados. A morfi
na deve
ser utilizada com cautela nos pacientes com asma brônquica, insu
ficiência hepática ou comprometimento da função renal.
6. Tolerância e dependência física. O uso repetido da morfina pro
duz tolerância aos seus efeitos depressor respiratório, analgésico,
eufórico e sedativo. No entanto, normalmente não se desenvolve
tolerância aos efeitos de constrição pupilar e de constipação. De
pendência física e psicológica ocorre rapidamente com a morfina e
com alguns dos outros agonistas (ver Figura 14.3). A retirada pro
duz umasérie de respostas autônomas, motoras e psicológicas que
incapacitam o indivíduo e causam sintomas graves (quase insupor
táveis). Entretanto, é muito raro que os efeitos sejam tão intensos a
ponto de levar a óbito. (Nota: a desintoxicação de indivíduos depen
dentes de morfina normalmente é alcançada pela administração via
oral de metadona, buprenorfina [ver adiante] ou clonidina.)
7. Interações farmacológicas. As interações de fármacos com morfi
na parecem ser raras, contudo as ações depressivas da morfi
na são
acentuadas pelas fenotiazinas, pelos inibidores da monoamina oxida
se (1MAO) e pelos antidepressivos tricíclicos (ver Figura 14.6).
B. Meperidina
A meperidina é um opioide sintético estruturalmente não relacionado à
morfi
na. Ela é utilizada contra a dor aguda.
1. Mecanismo de ação. A meperidi
na se liga aos receptores de opioi
des, particularmente aos receptores µ. Também se liga de modo
eficaz aos receptores K.
2. Ações. A meperidina causa depressão da respiração de forma se
melhante à morfina, mas não apresenta ação cardiovascular signifi
cativa quando administrada por via oral. Na administração IV, a me
peridina produz diminuição na resistência periférica e aumento no
fluxo sanguíneo periférico e pode aumentar a frequência cardíaca.
Como ocorre com a morfi
na, a meperidina dilata os vasos cerebrais,
aumenta a pressão do líquido cerebrospinal e contrai a musculatura
lisa (esse último menos intensamente do que a morfina). A meperi
dina não causa pupila puntiforme, mas dilatação das pupilas devido
a uma ação anticolinérgica.
3. Utilização terapêutica. A meperidina produz analgesia, mas não
é recomendada para uso prolongado, porque seu metabólito ativo,
normeperidina tem propriedades neurotóxicas significativas. Ao
Farmacologia Ilustrada 173
Contraindicação
absoluta para
meperidina e contraindicação
relativa para outros
analgésicos
narcóticos devido à
alta incidência de
coma hiperpiréxico
Maior depressão
do SNC,
particularmente
depressão
respiratória
Inibidores Sedativos-
da MAO -hi nóticos
Antidepressivos Fármacos
tricíclicos neurolépticos
Maiorsedação;efeitosvariáveis
sobreadepressãorespiratória
Figura 14.6
1nteração de fármacos com analgési
cos narcóticos. SNC = sistema nervoso
central; MAO = monoamina oxidase.
Legenda
Morfina
Meperidina
Fentanila
Figura 14.7
Tempoparaopicodeefeito
Duraçãodeação
15 minutos
2 a 4 horas
5 minutos
15 a 30 minutos
Tempo para o pico de efeito e duração
de ação de vários opioides administra
dos porvia IV.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-183-320.jpg)










![184 Clark, Finkel, Rey & Whalen
Chave: Nomsdofármaco
Inicialmenteconsidere com base nas
características do paciente, no diagnóstico
e sintomase leveem consideração
problemas médicosconcorrentes.
EPILEPSIAPARCIAL
Parcial simples,
parcial complexa
com ou sem
generalização
secundária
Paciente idoso
Lamotrigina
Levetiracetam
Topiramato
Lamotrigina
1Nome do fármaco
J
Considere esta opçãose as
crises persistem ou se os
efeitosadversos do 1°fármaco
impedem o uso.
1Nome do fármaco1
Considereestaalternativa se as
crises persistem ou osefeitos
adversos impedem otratamento.
Diva/proex
Carbamazepina Gabapentina
---:>...... Oxcarbazepina
Fenitoina
Lacosamida
.
Pregabalina
········->.....
Zoni
samida
Tiagabina
1Estimulaçãovagai1
Considere quando aadesão aotratamento,
as interações farmacológicasou os efeitos
adversos impedem otratamento
farmacológico.
·······--->·..·!Estimulação vagai]........
---':>......1Gabapentina ]...._
_
__,
:>......(Carbamazepinal-·.....---':>·....1Estimulação vagait.......
EPILEPSIAGENERALIZADAPRIMÁRIA
Ausência
Mioclônica
Tônico-clônica
Estado
epilético
Divalproex
Lamotrigina
Divalproex
Levetiracetam
Lamotrigina
Levetiracetam
Topiramato
Benzo..
dlazeplnas
Fosfenitofna
SÍNDROMEEPILÉPTICA
Rolãndica benigna
Espasmos infantis
(síndrome de West)
Lennox-Gastaut
Figura 15.5
· Gabapentina
Lamotrigina
Corticotropina
Vigabatrina
Divalproex
Lamotrigina
Toplramato
----.>......[Topiramato ]...._
_
__,
>..····[ Etosuximida �...................................................................
_
_
____.
:>...... Lamotrigina
Topiramato
""- D
iva/proex
-
-
-
v
......,__
Z
o
-
n
-
i
s
_
a
_
m
_
id
-
a
-
--<
---:>······ El����a����nas....................................................................
-
-
-
-
-
-
-
-
-
-
-
-
-
-
---.,.,.-........[Estimulação vagai,.......
----.:>·····�Eléirllituratoi> ]···························································································································
Carbamazepína
·······->..···· Levetiracetam
Topiramato
Benzodiazepinas
·······--->..··· Diva/proex
Topiramato
1LevetiracetamJ
--->..···r---
R
_
uft
_
n
_
a
_
m
_
id
_
a
_
-<
V
igabatrina
Zoni
samida
. ---:>······������ina ····································································
->..···· Lamotrigina
Zonísamida
_
_
____.
""-...... Elenzodiazepinas .......-
-
----'
""- 1 1
v -
-
-
v·.... Estimulação vagair......
Felbamato
Indicações terapêuticas para os fármacos anticonvulsivantes. Benzodiazepinas = diazepam e /orazepam.
IV. SELEÇÃO DO FÁRMACO
A escolha do tratamento farmacológico se baseia no tipo específico de crise
a ser combatida, variáveis específicas do paciente (p. ex., idade, condições
mórbidas simultâneas, estilo de vida e preferências pessoais) e características
do fármaco, incluindo custos e interações com outros fármacos. Por exemplo,
ataque parcial de crises são tratadas com medicamentos diferentes dos utili
zados nas crises generalizadas primárias ainda que a relação de fármacos efi
cazes se sobreponha. Muitos fármacos anticonvulsivantes podem ter a mesma](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-194-320.jpg)









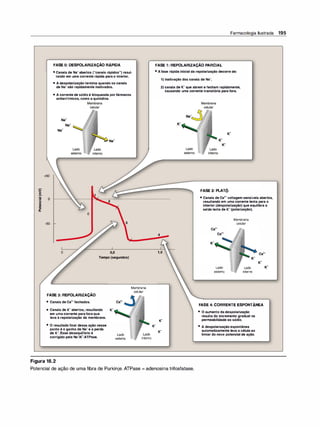






![202 Clark, Finkel,Rey& Whalen
oCORAÇÃO NORMAL
• Dentro de limites, quando o músculo cardíaco
é estirado, sua força de contração aumenta,
e, com isso, o débito cardíaco aumenta.
• Todavia, se o ventrículo for superestirado,
o efeito da contração ventricular diminui.
• O é o ponto normal de funcionamento
no coração sadio.
o
�
·-
2.
longo tempo), o potencial de repouso da membrana pode aumen
tar (- 70 mV em vez de - 90 mV), o que torna a membrana mais
excitável, aumentando o risco de arritmias (toxicidade).
b. Aumento da contratilidade do músculo cardíaco. A adminis
tração de glicosídeos digitálicos aumenta a força de contração
cardíaca, causando um débito cardíaco mais próximo ao do co
ração normal (Figura 16.9). Um aumento na contração do mio
cárdio leva à redução do volume diastólico final, aumentando,
dessa forma, a eficiência da contração (aumento da fração de
ejeção). A consequente melhora da circulação leva à redução
da atividade simpática, que, então, reduz a resistência periférica.
Juntos, esses efeitos causam reduçãoda frequência cardíaca. O
tônus vagai também é aumentado; então, a frequência cardíaca
diminui, e a demanda de oxigênio pelo miocárdio é reduzida. A
digoxina retarda a velocidade de condução através do nó AV, o
que é responsável pelo seu uso na fibrilação atrial. (Nota: no co
ração normal, o efeito inotrópico positivo dos glicosídeos digitáli
cos é neutralizado pelos reflexos autônomos compensatórios.)
Usos terapêuticos. O tratamento com digoxina está indicado em
pacientes com disfunção sistólicaventricularesquerdagrave depois
de iniciado o tratamento com inibidor da ECA e diurético. A digoxina
não está indicada em pacientes com IC diastólica ou direita. A prin-
o TRATAMENTO COM DIGOXINA
• A administração de digoxina desloca a curva
defunção ventricular para próximo do normal.
• A maior contratilidade ([3para (!]) leva ao
aumento do débito cardíaco.
• A diminuição dos reflexos simpáticos e do
tônus vascular causa diminuição na pressão
ventricular diastólica final ([il para �).
Normal
'O
8
o
�
--
--
-
-
--
rn
�
· -
.....
-
rn...t:
IC tratada com digioxina
Figura 16.9
-
:s
i!
Sintomasde �
débitoinadequado,
comofadiga
IC não tratada
Pressão ventricular diastólicafinal
INSUFICIÊNCIA CARDÍACA
DESCOMPENSADA
• Redução inicial da contratilidade ( rJpara 0)
devido à IC.
• Sintomas de débito cardíaco baixo se desenvolvem
- por exemplo, dispneia e edema.
Sintomas de pressão
venosa excessiva
� INSUFICIÊNCIA CARDÍACA
� COMPENSADA
• Aumento da pressão ventricular diastólica final
( m para [3) no esforço de manter um débito
cardíaco adequado.
• O aumento da pressão ventricular diastólica final
causa sintomas de congestão - por exemplo, dispneia.
Curvas de função ventricular no coração normal, na insuficiência cardíaca (IC) e na IC tratada com digoxina.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-212-320.jpg)





![208 Clark, Finkel, Rey & Whalen
Essaarritmiacomumenvolvefocosectópicos
múltiplosdecélulasatriais,criandoummovi
mentocaóticodeimpulsosatravésdoátrio.
Arespostaventricularérápida(100a150
batimentosporminuto)eirregular.Odébito
cardíacoestádiminuídoeaintolerânciaao
exercícioécomum.
Os�-bloqueadoressãousadosnafibrilação
atrialporquediminuemafrequênciacardíaca
epromovemaconversãoaoritmosinusal.
Otratamentodelongaduraçãoedosagem
baixacomanticoagulantereduzoriscode
ataquecardíacoqueestáassociadocoma
fibrilaçãoatrial.
FÁRMACOSANTIARRÍTMICOS
TIPODE
ARRITMIA
ARRITMIAS
ATRIAIS
PALPITAÇÃO ..........
(FLU1TER)ATRIAL
FIBRILAÇÃO
ATRIAL
TAQUICARDIAS
SUPRAVENTRICULARES
REENTRADANO
NÓAV ......
,__
Classe1
Quinidina
Classli ClasseIli
Propranolol
Amlodarona
Propranolol
Dofetilida
Propranolol
ClasseIV Outros
Verapamll ··· ·· ,_
[_
o
_
;
g
_
o
_
'}(
_
i
n
_
a
__,
]
.......[.....
_
0
1
_
·
1t
_
i
a
z
_
em
_
_,}......... Tratamento
antlcoagulante
.. · .....· ·
[ Digoxina
]
Acondução
éretardada
atravésdo
nóAV
com
propranolol,
verapamíl
oudigoxina.
TAQUICARDIA .
'-
SUPRAVENTRICULAR Adenosina
·······································································································� ll'
era,,amil �-·········
AGUDA
TAQUICARDIAS
VENTRICULARES
TAQUICARDIA
VENTRICULAR
AGUDA
FIBRILAÇÃO
VENTRICULAR
(quenãoresponde
à desfibrilaçãoelétrica)
Lldocafna
...........[ Lidocaína J.....................................
�
-
-
-
�
Amlodarona
Amlodarona
Essaarritmiaéacausacomumdemorteempacientesquetiveram
infartodomiocárdio.Odébitocardíacoestácomprometido,ea
taquicardiapodeevoluirparafibrilaçãoventricular.Portanto,a
taquicardiaventricularexigecuidadoimediato.
Figura 17.2
Legenda:
Eplnefrlna
Nomedo fármaco Fármacocomumente
usado
!N
o
m
e d
o fármaco
: Fármacoalternativo
Indicações terapêuticas para algumas arritmias comumente encontradas. AV = atrioventricular.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-218-320.jpg)












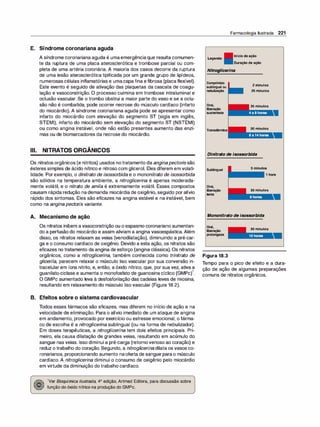

![contraindicados em pacientes com asma, diabetes, bradicardia grave, doença
vascular periférica e doença pulmonar obstrutiva crônica. (Nota: é importante
não interromper abruptamente o uso do [3-bloqueador. A dosagem deve ser re
duzidagradualmente por2 a 3semanas para evitar rebotes deangina, infarto do
miocárdio e hipertensão.)
V. BLOQUEADORES DE CANAIS DE CÁLCIO
O cálcio é essencial para a contração muscular. O influxo de cálcio aumenta na
isquemia devido à despolarização da membrana provocada pela hipoxia. Isso,
porsuavez, promove a atividade de várias enzimas que consomem trifosfatode
adenosina, esgotando, assim, os estoques de energia e agravando a isquemia.
Os bloqueadores de canais de cálcio protegem o tecido inibindo a entrada de
cálcio nas células cardíacas e musculares lisas dos leitos arteriais coronarianos
e sistêmicos.Todos os bloqueadores de canais de cálcio são, portanto, vasodi
latadores arteriolares que causam diminuição do tônus dos músculos lisos e da
resistência vascular. (Ver na p. 236 a descrição do mecanismo de ação desse
grupodefármacos.) Em doses clínicas, essesfármacos afetam primariamente a
resistênciade músculos lisos arteriolares coronarianos e periféricos. Seu uso no
tratamento da angina de esforço depende da redução do consumo de oxigênio
pelo miocárdio resultante da diminuição da pós-carga. Sua eficácia na angina
vasoespástica é devido ao relaxamento das artérias coronárias. (Nota: o vera
pamilafeta principalmente o miocárdio, e o nifedipi
no exerce maior efeito nos
músculos lisos nos vasos periféricos. O diltiazemapresenta ação intermediária.)
Todos os bloqueadores de canais de cálcio reduzem a pressão arterial. Eles
podem agravar a insuficiência cardíaca devido ao seu efeito inotrópico negativo.
(Nota: a angina variante causada por espasmo coronariano espontâneo, seja
em trabalho ou repouso [Figura 18.4] e não por aumento da demanda de oxi
gênio pelo miocárdio, é controlada pelos nitratos orgânicos ou bloqueadores de
canais de cálcio. Os [3-bloqueadores são contraindicados.)
A. Nifedipino
O nifedi
pino, um derivado da di-hidropiridina, funciona principalmente
como vasodilatador arteriolar. Esse fármaco apresenta efeito mínimo na
condução e na frequência cardíacas. O nifedipino é administrado por via
oral, normalmente naforma de comprimidos de liberação prolongada. Ele
sofre biotransformação hepática em produtos que são eliminados com
a urina e fezes. O efeito vasodilatador do nifedipino é útil no tratamento
da angina de Prinzmetal causada por espasmo coronariano espontâneo.
O nifedi
pino pode causar rubor, cefaleia, hipotensão e edema periféri
co como efeitos adversos decorrentes de sua atividade vasodilatadora.
Como com todos os bloqueadores de canais de cálcio, a constipação
é um problema. Como apresenta pouca ou nenhuma ação antagonista
simpática, o nifedi
pinopode causartaquicardia reflexa se avasodilatação
periférica é intensa. (Nota: o consenso geral é que as di-hidropiridinas de
ação curta devem ser evitadas na doença arterial coronariana devido a
evidências de aumento de mortalidade após infarto do miocárdio e au
mento de infartos do miocárdio agudo em pacientes hipertensos.)
B. Verapamil
A difenil-alquilamina verapamil reduz diretamente a velocidade de con
dução atrioventricular (AV) cardíaca, diminui a frequência e contratilidade
cardíaca, a pressão arterial e a demanda de oxigênio. O verapamilcausa
maior efeito inotrópico negativo do que o nifedi
pino, mas é um vasodilata
dor mais fraco. Como é extensamente biotransformado pelo fígado, deve
-se ter cuidado em ajustar a dosagem em pacientes com disfunção hepá
tica. O verapamilé contraindicado em pacientes com depressão da função
Farmacologia Ilustrada 223](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-233-320.jpg)
![224 Clark, Finkel, Rey & Whalen
DOENÇA CONCOMITANTE FÁRMACOS NORMALMENTE USADOS
NO TRATAMENTO DA ANGINA
NENHUMA
,
INFARTODOMIOCARDIO
RECENTE
ASMA,DPOC
HIPERTENSÃO
DIABETES
DOENÇA
-
RENALCRONICA
Figura 18.5
Nitratosdeação longa
Nitratosdeação longa
Nitratosdeação longa
Nitratosdeaçãolonga
Nitratosdeação longa
Nitratosdeação longa
[
Legenda:
13-Bloqueadores
13-Bloqueadores
JJ-Bloqueadores
13-Bloqueadores ]
Classedofármaco
Bloqueadores
decanais deca2•
Bloqueadores
decanais deca2•
Bloqueadores
decanais deca2•
Bloqueadores
de canais deca2•
Bloqueadores
de canais deca2•
[Classe do fármaco]
Fármacoscomumenteusados Fármacosmenoseficazes
Tratamento da angina em pacientes com doenças concomitantes. DPOC = doença pulmonar obstrutiva crônica.
cardíaca preexistente ou anormalidades de conduçãoAV. Ele também cau
saconstipação. O verapamildeve ser usado com cautela em pacientes que
estão tomando digoxina, pois ele aumenta os níveis desse fármaco.
C. Diltiazem
O diltiazem apresenta efeitos cardiovasculares que são similares aos do
verapamil. Ambos os fármacos diminuem a velocidade de condução AV e
reduzem ataxa de disparos do nó marca-passo sinusal.O diltiazem reduz
a frequência cardíaca, embora em menor extensão do que o verapamil,
e também diminui a pressão arterial. Além disso, o diltiazem pode aliviar
o espasmo coronariano e, portanto, é particularmente útil em pacientes
com angina variante. Ele é extensamente biotransformado nofígado.A in
cidência de efeitos adversos é baixa (a mesmados demais bloqueadores
de canais de cálcio). As interações com outros fármacos são as mesmas
indicadas para o verapamil.
,
VI. BLOQUEADORES DE CANAIS DE SODIO
A. Ranolazina
A ranolazina inibe a últimafase dacorrente de sódio (INatardia) melhoran
do a equação suprimento-e-demanda de oxigênio. A inibição da INatardia
reduz a sobre-carga intracelular de sódio e cálcio, melhorando, assim, a
disfunção diastólica. A ranozalinaé indicada para o tratamento da angina
crônica e pode ser usada só ou em combinação com outros tratamentos
tradicionais, mas, mais frequentemente, é usada como opção nos pacien
tes em que outros tratamentos anginosos falharam. A ranozalina não é
usada no tratamento de ataque agudo de angina.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-234-320.jpg)



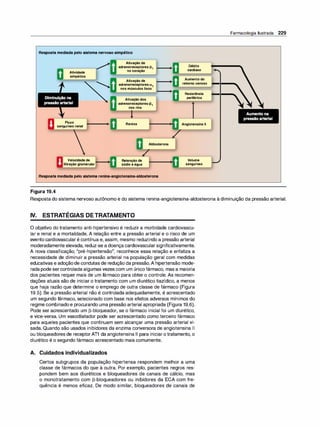
![230 Clark, Finkel, Rey & Whalen
�
DOENÇA
CONCOMITANTE CLASSESDEFÁRMACOSINDICADOSNOTRATAMENTODAHIPERTENSÃO
ANGINA PECTOR/S
[ Diuréticos
l [3-Bloqueadores Inibidores da ECA Bloqueadores
- de canais de ca2+
DEALTORISCO
- DIABETES
1 Diuréticos
1 [[3-Bloqueadores ] Inibidores da ECA
1 BRAs
1
Bloqueadores
de canais de ca2+
,__ AVERECORRENTE [ Diuréticos
l Inibidores da ECA
1 1 1 1
INSUFICIENCIACARDÍACA Diuré
t icos [3-Bloqueadores Inibidores da ECA BRAs
Antagonistas de
,__ receptor de
aldosterona
INFARTODO Antagonistas de
,__
MIOCÁRDIOPRÉVIO [3-Bloqueadores Inibidores da ECA receptor de
aldosterona
'-
DOENÇA
RENALCRÔNICA Inibidores da ECA
1 BRAs
1
Figura 19.5
Tratamento da hipertensão em pacientes com doenças concomitantes. As classes de fármacos em caixas azuis asse
guram melhor resultado (p. ex., diabetes ou doença renal) independente da pressão arterial. (Nota: os bloqueadores
de receptores de angiotensina [SRA] são uma alternativa aos inibidores da enzima conversora de angiotensina [ECA].
AVE = acidente vascularencefálico.
Angina
Diabetes
Hiperlipidemia
Insuficiência
cardíaca
Infartodo
miocárdioprévio
Alteraçãorenal
Asma
Figura 19.6
o 5 10 15
Porcentagemdepacientes
hipertensoscomadoença
concomitanteindicada
Frequência de doenças concomitantes
entre a população de pacientes hiper
tensos.
cálcio, inibidores da ECA e diuréticos são apropriados para o tratamento
da hipertensão em pacientes idosos, ao passo que os 13-bloqueadores e
os a-antagonistas são menos tolerados. Além disso, a hipertensão pode
coexistir com outras doenças que podem ser agravadas por alguns dos
fármacos anti-hipertensivos. Nesses casos, é importante encontraro me
lhorfármaco anti-hipertensivo para cada paciente em particular. A Figura
19.5 mostra o tratamento preferido em pacientes hipertensos com várias
doenças concomitantes, e a Figura 1 9.6 mostra a frequência de doenças
concomitantes na população de pacientes hipertensos.
B. Adesão do paciente ao tratamento anti-hipertensivo
A falta de adesão do paciente é a causa mais comum para a falha do
tratamento anti-hipertensivo. O paciente hipertenso normalmente é assin
tomático e é diagnosticado por triagem de rotina, antes da ocorrência de
lesão óbvia sobre um órgão-alvo. Dessa forma, o tratamento em geral é
direcionado para evitar as sequelas futuras da doença, em vez de aliviar
algum desconforto atual no paciente. Os efeitos adversos associados ao
tratamento anti-hipertensivo podem influenciar mais os pacientes do que
as vantagens futuras. Por exemplo, os 13-bloqueadores podem diminuir a
libido e induzirdisfunções eréteis em homens, particularmente os de meia
-idade e idosos. Essa disfunção sexual induzida pelo fármaco pode levar
o paciente a interromper o tratamento. Assim, é importante aumentar a
adesão do paciente selecionando cuidadosamente o regime de fármacos
que reduz osefeitos adversos e minimiza o número de dosificações neces
sárias cadadia. A associação de dois ou trêsfármacos em um comprimido
único, em associação dose-fixa, revelou melhorar a adesão ao tratamento
e o número de pacientes que alcançaram a pressão arterial desejada. A
Figura 19.7 mostra as normas do tratamento contra a hipertensão.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-240-320.jpg)
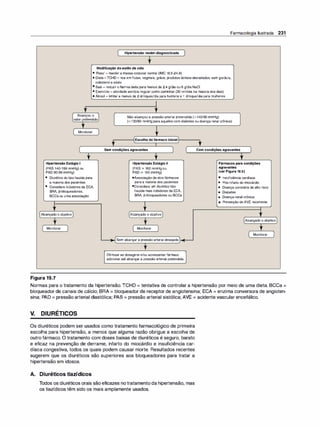

![VI. AGENTES BLOQUEADORES DE
P-ADRENORRECEPTORES
Atualmente os J3-bloqueadores são recomendados como tratamento de
primeira escolha contra a hipertensão quando está presente umadoença
concomitante (ver Fig. 19.5), por exemplo, em pacientes com infarto do
miocárdio ou com infarto prévio. Esses fármacos são eficazes, mas apre
sentam algumas contraindicações.
A. Ações
Os J3-bloqueadores reduzem a pressão arterial primariamente pela dimi
nuição do débito cardíaco (Figura 1 9.9). Eles também podem diminuir o
efluxo simpático do SNC e inibira liberação de reninados rins, reduzindo,
assim, a formação de angiotensina li e a secreção de aldosterona. O pro
tótipo dos J3-bloqueadores é o propranolo/, que atua em receptores-J31 e
J32• Bloqueadores seletivos de receptores J31, como metoprolole atenolol,
estão entre os J3-bloqueadores mais comumente prescritos. O nebivololé
um bloqueador seletivo de receptores J31 que aumenta também a produ
ção de óxido nítrico, levando à vasodilatação. Os J3-bloqueadores seleti
vos devem ser administrados cautelosamente em pacientes hipertensos
que também têm asma. Os J3-bloqueadores não seletivos, como propra
nolol e nado/o/, são contraindicados devido ao bloqueio da broncodilata
ção mediada por J32• (Ver p. 222 para a discussão sobre J3-bloqueadores.)
Os J3-bloqueadores devem ser usados com cautela no tratamento de pa
cientes com insuficiência cardíaca aguda ou doença vascular periférica.
B. Usos terapêuticos
1. Subgrupos de população hipertensa. Os J3-bloqueadores são mais
eficazes no tratamento de pacientes brancos do que de negros, sen
do também mais eficazes em pacientes jovens do que em idosos.
(Nota: condições que desaconselham o uso de J3-bloqueadores [p. ex.,
doença pulmonar obstrutiva crônica grave, insuficiência cardíaca con
gestiva crônicaedoençavascularperiféricaoclusivasintomáticagrave]
são mais comumente encontradas em pacientes idosos e diabéticos.)
2. Pacientes hipertensos com doenças concomitantes. Os J3-blo
queadores são úteis no tratamento de condições que podem coexis-
Bloqueadoresde
p-adrenorreceptores
Figura 19.9
Ativaçãode
adrenorreceptores
131 nocoração Débitocardíaco
Resistência
periférica
t
a Renina l Angiotensinali
[J
D Aldosterona
__,
J/
Retençãode
sódioeágua -----+[)
Volume
sanguíneo
Efeitos dos fármacos bloqueadores dos J3-adrenorreceptores.
Farmacologia Ilustrada 233](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-243-320.jpg)


![236 Clark, Finkel, Rey & Whalen
Ir.ti Dilataçã
_
o de vasos
W coronar1anos
Nifedipino 1
Verapamil 1
Diltiazem 1
1
1
Ação Ação
fraca forte
m Condução AV
Poucoefeito
Nifedipino ]�
Verapamil 1
'-----
;:::::!
Diltiazem 1
.____.
Diminuída Aumentada
f'!!I Frequência de
.:.1 efeitos adversos
Nifedipino 1 1 18°/o
:=:
=
=
=
==:-
-
�
Verapamil ._
l_
_
_
__,
I 9°/o
Diltiazem D 2°/o
Infrequente
Figura 19.13
Frequente
Ações dos bloqueadores de canais de
cálcio. AV = atrioventricular.
reduzindo, assim, a pressão arterial e diminuindo a retenção de sal e água.
Os BRAs não aumentam os níveis de bradicinina. Eles diminuem a nefro
toxicidade do diabetes, sendo, assim, um tratamento atraente a diabéticos
hipertensos. Seus efeitos adversos são semelhantes aos dos inibidores da
ECA, embora o risco de tosse e angioedema seja significativamente menor.
Os BRAs também são fetotóxicos e não devem ser usados por mulheres ges
tantes. (Nota: os BRAs são discutidos no Capítulo 16.)
IX. INIBIDORES DE RENINA
O inibidor seletivo de renina, ali
squireno, está disponível para o tratamento da
hipertensão. O a/isquireno inibe diretamente a renina e, assim, atua mais pre
cocemente no sistema renina-angiotensina-aldosterona do que os inibidores
de ECA ou BRAs. Ele reduz a pressão arterial com eficácia similar aos BRAs,
inibidores da ECA e tiazidas. O alisquireno pode ser associado com outros
anti-hipertensivos, como diuréticos, inibidores da ECA, BRAs ou bloqueado
res de canais de cálcio. O alisquireno pode causar diarreia, em especial em
dosagens elevadas. Ele também pode causar tosse e angioedema, mas pro
vavelmente com menos frequência do que os inibidores da ECA. Como os ini
bidores da ECA e os BRA, o a/iquireno é contraindicado durante a gestação.
O alisquireno está disponível em associação de doses fixas com valsartana,
bem como com hidroclortiazida. A hiperpotassemiafoi significativamente mais
comum em pacientes que receberam alisquireno e valsartana. O aliquireno é
biotransformado pela CIP3A4 e é sujeito a interações de fármacos.
X. BLOQUEADORES DE CANAIS DE CÁLCIO
Os bloqueadores de canais de cálcio são recomendados quando os fárma
cos de primeira escolha estão contraindicados ou são ineficazes. Eles são
eficazes no tratamento da hipertensão em pacientes com angina ou diabetes.
Doses elevadas de bloqueadores de canais de cálcio de curtaduração devem
ser evitadas devido ao maior risco de infarto do miocárdio por vasodilatação
excessiva e acentuada estimulação cardíaca reflexa.
A. Classes de bloqueadores de canais de cálcio
Os bloqueadores de canais de cálcio são divididos em três classes quími
cas, cada uma com propriedades farmacocinéticas e indicações clínicas
diferentes (Figura 19.13).
1. Difenilalquilaminas. O verapamil é o único representante dessa
classe aprovado nos Estados Unidos atualmente. O verapamil é o
menos seletivo dos bloqueadores de canais de cálcio e apresenta
efeitos significativos nas células cardíacas e do músculo liso vascu
lar. Ele é usado também notratamento da angina, das taquiarritmias
supraventriculares e para prevenir a enxaqueca e a cefaleia em sal
vas. Bloqueio atrioventricular de primeiro grau e constipação são
efeitos adversos dose-dependentes do verapamil.
2. Benzotiazepina. O diltiazem é o único membro dessa classe que
está aprovado nos Estados Unidos atualmente. Como o verapamil,
o dilti
azem afeta tanto as células cardíacas quanto do músculo liso
vascular, mas apresenta efeito inotrópico cardíaco negativo menos
pronunciado comparado ao efeito do verapamil. O diltiazem apre
senta um perfil de efeitos adversos favorável.
3. Di-hidropiridinas. Essaclasse de bloqueadores de canais de cálcio
em rápida expansão inclui o nifedipino de primeira geração e cinco](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-246-320.jpg)



![240 Clark, Finkel, Rey & Whalen
Legenda:
Nitroprussiato �
Labeta/oi
Feno/dopam
Nicardipino
Figura 19.16
Início de ação
Duração de ação
Imediato
1 a 2 minutos
5 a 1O minutos
2 a 5 minutos
5a 1O minutos
Tempo até alcançar o pico de efeito e
a duração de ação de alguns fármacos
usados na emergência hipertensiva.
dem ser graves e exigir o uso simultâneo de um diurético de alça e um
�-bloqueador. O minoxidil causa séria retenção de sódio e água, acarre
tando sobrecarga de volume, edema e insuficiência cardíaca congestiva.
(Nota: otratamento com minoxidiltambém causa hipertricose [crescimen
to dos pelos do corpo]. Atualmente, esse fármaco é usado topicamente
no tratamento da calvície masculina.)
XV. EMERGÊNCIA HIPERTENSIVA
A emergência hipertensiva é uma situação rara, mas ameaçadora à sobrevi
vência, na qual a pressão arterial diastólica é maior do que 150 mmHg (com
pressão arterial sistólica acima de 21O mmHg) em uma pessoa de outra for
ma saudável ou uma pressão arterial diastólica maior que 130 mmHg em um
indivíduo com complicações preexistentes, como encefalopatia, hemorragia
cerebral, insuficiência ventricular esquerda ou estenose aórtica. O objetivo
terapêutico é a rápida redução da pressão arterial.
A. Nitroprussiato de sódio
O nitroprussiato é administrado por via IV e causa vasodilatação rápida
com taquicardia reflexa. Ele é capaz de reduzir a pressão arterial em to
dos os pacientes independente da causade hipertensão (Figura 19.16). O
fármaco apresenta poucos efeitosforadosistemavascular, atuando igual
mente nos músculos lisos arteriais e venosos. (Nota: como o nitroprus
siato também atua nas veias pode reduzir a pré-carga cardíaca.) O nitro
prussiatoé rapidamente biotransformado (meia-vida de minutos) e requer
infusão contínua para manter sua ação anti-hipertensiva. O nitroprussiato
de sódio causa poucos efeitos adversos, exceto a hipotensão causada
por dosagem excessiva. A biotransformação do nitroprussiato resulta na
produção de íon cianeto. Embora a toxicidade por cianeto seja rara, ela
pode sertratadacom a infusão de tiossulfato de sódio para produzirtiocia
nato, que é menos tóxico e é eliminado pelos rins. (Nota: o nitroprussiato
é tóxico se administrado por via oral devido à sua hidrólise a cianeto.) O
nitroprussiato é sensível à luz e, em solução, deve ser protegido da luz.
B. Labetalol
Labetalol é um bloqueador a e � e é administrado em bolus ou infusão
porvia IV nas emergências hipertensivas. O labetalolnão causa taquicar
dia reflexa. Ele apresenta a contraindicação de ser um bloqueador não
seletivo. A principal limitação é a longa meia-vida, o que impede a titula
ção rápida (ver Figura 19.16).
C. Fenoldopam
Feno/dopam é um agonista periférico de receptor de dopamina1 admi
nistrado por infusão IV. Diferentemente dos outros anti-hipertensivos pa
renterais, o feno/dopam mantém ou aumenta a perfusão renal, enquanto
diminui a pressão arterial. O fenoldopan relaxa principalmente os vasos
renais (artéria renal, arteríolas aferente e eferente) e mesentéricos arte
riais, com ação vasodilatadora menor nas artérias coronárias e cerebrais
e nas veias (vasos de capacitância). A ação diurética do feno/dopam é
causada principalmente pelo aumento do fluxo de sangue renal. O feno/
dopam pode ser usado com segurança em todas as emergências hiper
tensivas e pode ser particularmente benéfico em pacientes com insufi
ciência renal. O fármaco é contraindicado em pacientes com glaucoma.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-250-320.jpg)




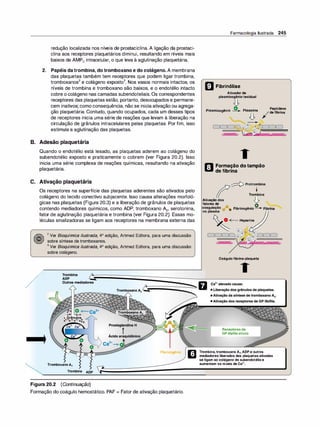






![252 Clark, Finkel, Rey & Whalen
ft Heparina
W nãofracionada
Heparina
J
Antitrombina
IFllft©Jll' �(!
� <J
OOo / ,P
!
� �()
v
hr-
-....
l A hepar
inanão
fracionadaacelera
a interação da
antitrombina com
atrombinae o
FatorXa.
°oº rY
__, Ooocoo<P°
A interaçãodaantitrombinacom a
heparinaea HBMMé mediada por
umasequência de pentassacarídeos.
/º
�w�ooc>ooOc;O
�D
Trombina
m
Antitrombina
HBMM
�
IF�©ll' �� HBMMacelera
HBMM
@ � <J
seletivamente a
o
I
()
interação da
antitrombina
l l com o FatorXa.
.. ..
Figura 20.14
A heparina e a heparina de baixa massa molecular (HBMM) intermedeiam a inativação da trombina ou do FatorXa.
Meia-vida
intravenosa
Resposta
anticoagulante
Biodisponibilidade
Principais efeitos
adversos
Local de
tratamento
Figura 20.15
2 horas 4 horas
Variável Previsível
200/o 90%
Sangramentos Sangramentos
frequentes menosfrequentes
Hospital Hospital e
paciente externo
Algumas propriedades da heparina e
das heparinas de baixa massa molecu
lar(HBMM).
20.12). (Nota: nesta discussão, o termo heparina indica a forma não fracio
nada do fármaco.) O conhecimento de que formas de heparina de baixa
massa molecular(HBMM) também conseguem atuarcomo anticoagulantes
levou ao isolamento da enoxaparina, a primeira HBMM(< 6.000) disponível
nos Estados Unidos. As HBMMs são compostos heterogêneos (elas têm
um terço do tamanho da heparina não fracionada) produzidos pela despo
limerização química ou enzimática da heparina não fracionada. Como são
isentas de desvantagens associadas ao polímero, estão substituindo o uso
da heparinaem várias situações clínicas.A heparina é usada na prevenção
de trombose venosa e no tratamento de uma variedade de doenças trom
bóticas, como embolia pulmonar e infarto agudo do miocárdio.
1. Mecanismo de ação. A heparina atua em inúmeros alvos molecula
res, mas seu efeito anticoagulante é consequência da ligação à anti
trombina Ili, com a rápida inativação subsequente dos fatores de coa
gulação (Figura 20.13). A antitrombina Ili é uma a-globulina e inibe
serina-proteases, incluindo vários dos fatores de coagulação- sendo
os mais importantes a trombina (Fator lla) e o Fator Xa (ver Figura
20.1O). Na ausência de heparina, a antitrombina Ili interage lentamen
te com a trombina e o FatorXa. As moléculas de heparina se ligam à
antitrombina Ili, induzindo alteração conformacional que acelera sua
velocidade de ação cerca de mil vezes. A heparina também serve de
molde catalítico para a interação da antitrombina Ili e os fatores de
coagulação ativados. Ela atua comoverdadeiro catalisador, permitindo
que a antitrombina Ili se combine rapidamente com a trombina circu
lante e o Fator Xa e os iniba (Figura 20.14). Em contraste, as HBMMs
complexam com a antitrombina Ili e inativam o Fator Xa (incluindo o
localizado nassuperfíciesdas plaquetas), mas não se ligam tão avida
mente à trombina. Sem dúvida, a ativação das plaquetas em repouso
é menos provável com as HBMMs do que com a heparina. (Nota: a
sequência de pentassacarídeo presente na heparina e nas HBMMs
permite sua ligação à antitrombina Ili [ver Figura 20.14].)
2. Usos terapêuticos. A heparina e as HBMMs limitam a expansão
dos trombos evitando a formação de fibrina. A heparina é o prin
cipal fármaco antitrombótico para o tratamento da trombose aguda
de veias profundas e do embolismo pulmonar. A incidência de epi-](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-262-320.jpg)









![262 Clark, Finkel, Rey & Whalen
TRATAMENTO
MEDICAMENTOSO
•Tratamento com fármacos
quesão inibidores da
di-hidrofolato-redutase.
MÁABSORÇÃO
• Patologia do intestino
delgado.
• Alcoolismo.
DEFICIÊNCIASNADIETA
• Durantea gestação.
• Durantea lactação.
�
DEFICIÊNCIA
DE FOLATO
Biossíntese de
aminoácidos
Síntesede
purinas
Síntesede
pirimidinas
Síntese de
DNAe RNA
Anemia
megaloblástica
Figura 20.27
Causas e consequências da falta de
ácido fálico.
A. Ferro
O ferro é armazenado nas células da mucosa intestinal comoferritina (um
complexo ferroproteína) até que ele seja necessário para o organismo. A
deficiência de ferro resulta de perdas de sangue agudas ou crônicas, da
ingestão insuficiente durante períodos de crescimento rápido em crian
ças ou em mulheres grávidas ou com menstruação intensa. Assim, a defi
ciência de ferro resulta de um equilíbrio negativo decorrente da perda dos
estoques de ferro e/ou da ingestão inadequada, culminando em anemia
microcítica hipocrômica (eritrócitos de tamanho pequeno e com pouco
ferro). A suplementação de sulfato ferroso é necessária para corrigir a
deficiência. Distúrbios gastrintestinais causados por irritação local são os
efeitos adversos mais comuns da suplementação de ferro, quando po
dem ser usadas formulações parenterais de ferro, como o ferrodextrano.
B. Ácido fólico (folato)
O uso primário do ácido fólicoéo tratamento de estadosdeficitários que se
originam da ingestão inadequada da vitamina. A deficiência de folato pode
ser causada por: 1) aumento da demanda (p. ex., gestação e lactação); 2)
baixa absorção causada por patologia do intestino delgado; 3) alcoolismo
ou 4) tratamento com fármacos que são inibidores da di-hidrofolato-redu
tase (p. ex., metotrexato, pirimetamina e trimetoprima) em cujo caso deve
ser usada a forma reduzida ou ativa da vitamina (ácido folínico, também
denominado leucovorin cálcico). O resultado primárioda deficiência de áci
do fólico é a anemia megaloblástica (eritrócitos de grande tamanho), que
é causada pela diminuição da síntese de purinas e pirimidinas. Isso leva
à incapacidade do tecido eritropoiético de produzir DNA e, dessa forma,
proliferar (Figura 20.27). (Nota: para evitar complicações neurológicas da
deficiência de vitamina 812, é importante avaliara base da anemia megalo
blástica antes de instituir o tratamento.Tanto a deficiência de vitamina 812
como a de to/ato podem causar sintomas similares [ver adiante].) O ácido
fó
lico é bem absorvido no jejuno, a menos que esteja presente alguma
patologia. Se quantidades excessivas da vitamina são ingeridas, elas são
excretadas na urina e nas fezes. O ácido fólico administrado por via oral
não é tóxico. Não foram registrados efeitos adversos substanciais além de
raras reações de hipersensibilidade na injeção parenteral.
C. Cianocobalamina (vitamina 812)
Deficiências de cianocobalamina podem resultar de baixa oferta na dieta
ou, mais comum, de má absorção da vitamina devido à insuficiência das
células parietais gástricas de produzir fator intrínseco (como na anemia
perniciosa) ou à perda da atividade do receptor necessária para a absor
ção intestinal da vitamina. O fator intrínseco é uma glicoproteína produ
zida pelas células parietais do estômago e é necessário para absorção
da vitamina 812• Pacientes com cirurgia bariátrica (tratamento cirúrgico
GI contra obesidade) necessitam de suplementação de vitamina 812 em
dose elevada por via oral, sublingual ou, uma vez por mês, por via pa
renteral. Síndromes de má absorção inespecífica ou ressecção gástri
ca também podem causar deficiências de cianocobalamina. A vitamina
pode ser administrada oralmente (nas deficiências nutricionais) ou por
via IM ou SC profunda (contra a anemia perniciosa). (Nota: a administra
ção somente de ácido fólico reverte as anormalidades hematológicas e,
assim, mascara as deficiências de cianocobalamina, a qual pode, então,
evoluir para grave disfunção e doença neurológica. A causa da anemia
megaloblástica precisa ser determinada para ser específica em termos
de tratamento. Dessaforma, a anemia megaloblástica não deve ser trata
da apenas com ácido fólico, mas com a associação de to/ato e vitamina
812.) O tratamento deve continuar portodaa vida do paciente se ele sofre](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-272-320.jpg)



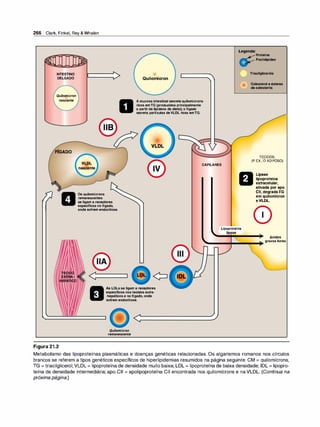
![Farmacologia Ilustrada 267
Tipo 1 (HIPERQUILOMICRONEMIA FAMILIAR)
• Hiperquilomicronemia massivaemjejum, mesmo após ingestão normal degorduras
nadieta, resultando em grandeelevação nos níveis plasmáticos deTG.
• Deficiência de lipoproteína lipaseou deapolipoproteína Cll normal (raro).
Quilomícron
• OTipo 1 nãoestáassociado a aumento de doenças coronarianas cardíacas.
•Tratamento: dieta pobre em gorduras. Nenhumtratamentofarmacológico éeficazcontra hiperlipidemiaTipo 1.
Tipo llA (HIPERCOLESTEROLEMIA FAMILIAR)
• LDL elevada com níveis normais deVLDL devido a um bloqueio nadegradação da LDL.
Isso resulta no aumento do colesterol plasmático, masem níveis normais deTG.
• Causada pordefeitos nasíntese ou no processamento dos receptores de LDL.
• Grande aceleração de doenças cardíacas isquêmicas.
•Tratamento: dieta. Heterozigotos: colestiraminae niacinaou uma estatina.
Tipo 118 (HIPERLIPIDEMIA FAMILIAR COMBINADA [MISTA])
• Semelhante aoTipo llA, exceto pelo fato de os níveis deVLDLtambémestarem
aumentados, resultandoem aumento dos níveis plasmáticos deTG ede colesterol.
•A causa é a produção excessivadeVLDL pelofígado.
• Relativamente comum.
•Tratamento: dieta.Tratamentofarmacológico semelhante ao doTipo llA.
Tipo Ili (DISBETALIPOPROTEINEMIA FAMILIAR)
• Concentraçõesséricas de IDL aumentadas, resultando em aumento dos níveis deTG ecolesterol.
• A causaé a produção excessiva ou a pouca utilização de IDL devido à apolipoproteína E mutante.
• Nos pacientes de meia idade desenvolvem-sexantomas edoenças vasculares aceleradas.
•Tratamento: dieta. Otratamentofarmacológico inclui niacinae fenofibratoou uma estatina.
Tipo IV (HIPERTRIGLICERIDEMIA FAMILIAR)
• Os níveis deVLDL estão aumentados, ao passo queos de LDL estão normais ou diminuídos,
resultando em níveis normais ou elevados de colesterol e níveis muitoelevados deTG circulante.
• A causaé a produção excessivae/ou a diminuição da remoção deTG daVLDL do soro.
• Éumadoença relativamente comum.Apresenta poucas manifestações clínicas além da
aceleração de doença cardíaca isquêmica. Pacientes portadores desse distúrbio
frequentementesão obesos, diabéticose hiperuricêmicos.
•Tratamento: dieta. Se necessário,o tratamentofarmacológico inclui ni
acinae/ou fenofibrato.
Tipo V (HIPERTRIGLICERIDEMIA FAMILIAR MISTA)
• Níveis séricos deVLDL e quilomícrons elevados. LDL normal ou reduzida. Isso resulta
naelevação dos níveis de colesterol e na grande elevação dos deTG.
•A causaé o aumento na produção ou a redução nadepuração daVLDL e dos
quilomícrons. Em geral, é um defeito genético.
• Ocorre mais comumenteem adultos obesose/ou diabéticos.
•Tratamento: dieta. Se necessário,o tratamentofarmacológico inclui ni
acinae/ou
fenofibratoou umaestatina.
Figura 21.2 (Continuação)
VLDL
VLDL
Quilomícron VLDL](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-277-320.jpg)
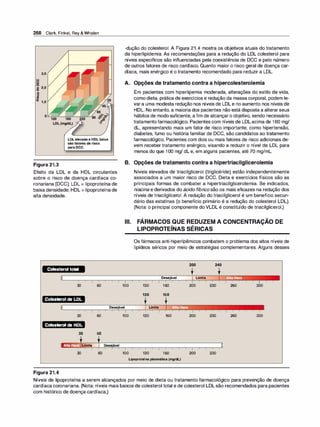

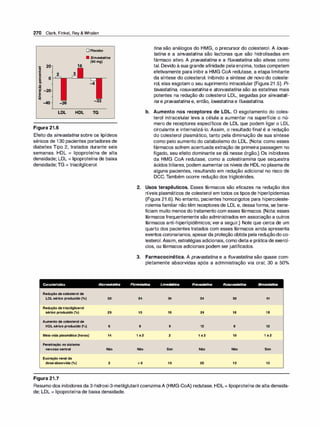

![272 Clark, Finkel, Rey & Whalen
300
Total
�
..J
32
CJI
.§. 200- LDL
-
e
J!I
li 100
-
o HDL
o
� . . . .
1 2 3 4
Meses
Figura 21.1O
. .
5 6
Níveis plasmáticos de colesterol em pa
cientes hiperlipêmicos durante o trata
mento com niacina. LDL = lipoproteína
de baixadensidade; HDL = lipoproteína
de alta densidade.
Quilomícron
TECIDOS
Capilares (p.ex., ADIPOSO)
�"""" Genfibrozila
Lipoproteína
lipase
IDL
Quilomícrons
remanescentes.......-
Figura 21.11
,
Acidos
graxos
livres
Ativação da lipoproteína lipase pelo
genfibrozila. VLDL = lipoproteína de
densidade muito baixa; IDL = lipopro
teína de densidade intermediária.
3.
no tratamento de outras hipercolesterolemias graves, frequentemen
te associadaa outros anti-hiperlipêmicos. Além disso, elaé ofármaco
anti-hiperlipidêmico mais potente para elevar os níveis plasmáticos
de HDL, que é a sua indicação de uso clínico mais comum.
Farmacocinética. A niaci
na é administrada por via oral. No organis
mo, ela é convertida em nicotinamida, que é incorporada ao cofator
nicotinamida adenina dinucleotídeo (NAD+). A niacina, o derivado
nicotinamida e outros metabólitos são excretados na urina. (Nota: a
nicotinamida isolada não reduz os níveis lipídicos plasmáticos.)
4. Efeitos adversos. Os efeitos adversos mais comuns da niacina são
intenso rubor cutâneo (acompanhado por uma desconfortável sen
sação de calor) e prurido. A administração de ácido acetilsalicílico
previamente à niacina reduz o rubor, que é mediado por prostaglan
dinas. A formulação de liberação lentada niaci
na, administrada uma
vez ao dia ao deitar, reduz os desconfortáveis efeitos adversos ini
ciais.Alguns pacientes também sentem náuseas e dor abdominal.A
niacina inibe a secreção tubular de ácido úrico, predispondo, assim,
à hiperuricemia e à gota.Também foram relatadas hepatotoxicidade
e intolerância à glicose.
C. Os fibratos: fenofibrato e genfibrozila
O fenofibrato e o genfibrozila são derivados do ácido fíbrico que reduzem
os triacilgliceróis e aumentam os níveis de HDL do plasma. Ambos têm
o mesmo mecanismo de ação. No entanto, o fenofibrato é mais eficaz do
que o genfibrozila na redução dos níveis plasmáticos de colesterol LDL
e triglicérides.
1. Mecanismo de ação. Os receptores ativados pelo proliferador de
peroxissomo (RAPPs) são membros da família de supergenes de
receptores nucleares que regula o metabolismo lipídico. Os RAPPs
funcionam como fatores de transcrição ativados pelo ligante. Eles
são ativados ao ligarem-se aos seus ligantes naturais (ácidos gra
xos ou eicosanoides) ou a fármacos hipolipidêmicos. A seguir, eles
se ligam aos elementos de resposta proliferadores de peroxissomo,
localizados em numerosos promotores de genes. Os RAPPs regu
lam, particularmente, a expressão de genes codificadores de proteí
nas envolvidas na estrutura e função das lipoproteínas.A expressão
de genes mediada por fibrato leva, em última instância, à redução
das concentrações de triacilglicerol pelo aumento da expressão da
lipoproteína lipase (Figura 21 .11) e pela diminuição da concentra
ção de apo C-11. Os fibratos também aumentam o nível de colesterol
HDL pelo aumento da expressão de apo AI e apo All. O fenofibrato
é um pró-fármaco que produz um metabólito ativo, o ácido fenofíbri
co, responsável por seus efeitos principais.
2. Usos terapêuticos. Os fibratos são utilizados no tratamento das hi
pertriacilglicerolemias, provocando uma diminuição significativa nos
níveis plasmáticos de triacilglicerol. O f
enofibrato e o genfibrozila são
particularmente úteis no tratamento da hiperlipemia Tipo Ili (disbeta
lipoproteinemia), na qual se acumulam as partículas lipoproteicas de
densidade intermediária. Pacientes portadores de hipertriacilglicero
lemia (doença Tipo IV [com elevada VLDL] ou Tipo V [com elevados
VLDLequilomícrons]) que não respondem àdieta eaoutrosfármacos
também podem se beneficiardotratamento com esses fármacos.
3. Farmacocinética. Ambos os fármacos são completamente absor
vidos após administração via oral. O genfibrozila e o fenofibrato se](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-282-320.jpg)





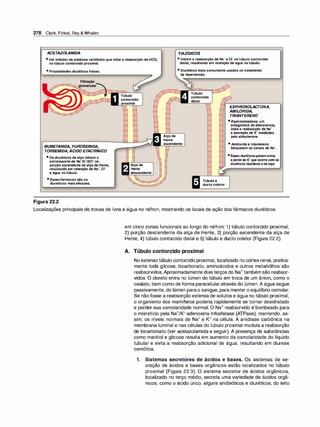








![4. Farmacocinética. A espironolactona é completamente absorvida
por via oral e é fortemente ligada às proteínas. Ela é rapidamente
convertida no metabólito ativo, a canrenona. A ação da espironolac
tona é amplamente decorrente do efeito da canrenona, que tem ati
vidade bloqueadora de mineralocorticoide. A espironolactona induz
o citocromo hepático P450.
5. Efeitos adversos. A espironolactona frequentemente causa distúr
bios gástricos, podendo causar úlcera péptica. Como se assemelha
quimicamente a alguns esteroides sexuais, a espironolactona pode
atuar em receptores em outros órgãos, induzindo ginecomastia em
homens e irregularidades menstruais em mulheres; portanto, o fár
maco não deve ser administrado de forma crônica em doses ele
vadas. A espironolactona é mais eficaz nos estados edematosos
leves, nos quais ela é administrada durante poucos dias. Em doses
baixas, ela pode ser usada cronicamente, com poucos efeitos ad
versos. Podem ocorrer hiperpotassemia, náuseas, letargia e confu
são mental.
B. Triantereno e amilorida
O triantereno e a amilorida bloqueiam os canais de transporte de Na+,
resultando em diminuição na troca Na+/K+. Embora apresentem ação diu
rética poupadora de K+ semelhante à da espironolactona, a capacidade
desses fármacos de bloquear os locais de troca Na+/K+ no túbulo coletor
não depende da presençade aldosterona. Dessa forma, eles apresentam
atividade mesmo em indivíduos com doença de Addison. Como a espiro
nolactona, o triantereno e a amilorida não são diuréticos muito eficientes.
Tanto o tri
antereno quanto a amilorida são frequentemente usados em
associação a outros diuréticos, em geral devido à sua propriedade pou
padora de potássio. Por exemplo, muito semelhante à espironolactona,
eles previnem a perdade K+que ocorre com os tiazídicos e a furosemida.
Os efeitos adversos do triantereno são cãibras nas pernas e o possível
aumento do nitrogênio ureico sanguíneo, bem como retenção de ácido
úrico e K+.
VII. INIBIDORES DA ANIDRASE CARBÔNICA
A acetazolamida inibe a enzima anidrase carbônica nas células epiteliais do
túbulo proximal. Os inibidores da anidrase carbônica são usados com mais
frequência porsuas outras ações farmacológicas do que por seu efeito diuré
tico, pois são muito menos eficazes do que os diuréticos tiazídicos ou de alça.
A. Acetazolamida
1 . Mecanismo de ação. A acetazolamida inibe a anidrase carbônica
localizada intracelularmente (citoplasma) e na membrana apical do
epitélio tubular proximal (Figura 22.9). (Nota: a anidrase carbônica
catalisaa reação entre C02 e H20, produzindo H2C03, que esponta
neamente se ioniza em H+ e HCO 3- [bicarbonato].) A diminuição na
capacidade detrocaentre Na+ por H+ na presença de acetazolamida
resulta em umadiurese leve.Além disso, o HC03-é retido no lúmen,
com acentuada elevação no pH urinário. A perda de HC03- causa
acidose metabólica hiperclorêmica e redução da eficácia diurética
após alguns dias de tratamento. As mudanças na composição de
eletrólitos urinários, induzidas pela acetazolamida, estão resumidas
na Figura 22.1O. A excreção de fosfato é aumentada por mecanismo
desconhecido.
Farmacologia Ilustrada 287
Lúmen
H20 + C02
___.________, t
Anidrase
carbônica
Célula epitelial
do túbulo renal
Figura 22.9
Sangue
Papel da anidrase carbônica na reten
ção de sódio pelas células epiteliais do
túbulo renal.
Figura 22.1O
HC0-
3
Volume
de urina
Mudanças relativas na composição da
urina induzidas pela acetazolamida.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-297-320.jpg)




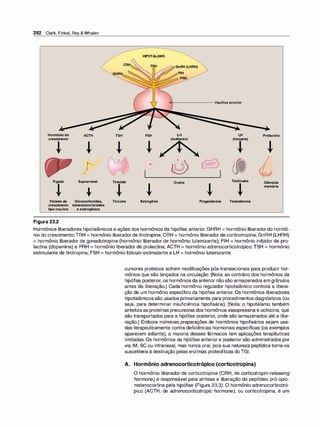
![produto do processamento pós-translacional desse polipeptídeo precur
sor. (Nota: o CRH é usado no diagnóstico para diferenciarentre síndrome
de Cushing e células ectópicas produtoras de ACTH.) Outros produtos
do pró-opiomelanocortina são o hormônio 'Y-melanócito estimulante e as
�-lipotropinas, precursoras das endorfinas. Normalmente, o ACTH é libe
rado da hipófise em pulsos com um predomínio de ritmo diurno, a maior
concentração acontecendo aproximadamente às 6 horas e a menor ao
anoitecer. O estresse estimula a secreção, e o cortisol suprime sua libe
ração, atuando via retroalimentação negativa.
1 . Mecanismo de ação. O órgão-alvo do ACTH é a suprarrenal, onde
ele se liga a receptores específicos na superfície celular. O receptor
ocupado ativa processos acoplados à proteína G para aumentar o
AMPe• que, por sua vez, estimula a etapa limitante da síntese do
adrenocorticosteroide (de colesterol a pregnenolona). Essa via ter
mina com a síntese e a liberação dos adrenocorticosteroides e dos
androgênios suprarrenais (ver Figura 23.3).
2. Usos terapêuticos. A disponibilidade de adrenocorticosteroides
sintéticos com propriedades específicas limitou o uso da corticotro
pina, principalmente como ferramenta diagnóstica para diferenciar
entre insuficiência suprarrenal primária (doença de Addison asso
ciada à atrofia suprarrenal) e insuficiência suprarrenal secundária
(causada pela secreção inadequada de ACTH pela hipófise). Pre
parações de corticotropina terapêuticas são extraídas da hipófise
anterior de animais domésticos ou ACTH humano sintético. Este
último, a cosintropina, que consiste nos 24 aminoácidos aminoter
minais do hormônio, é preferido para o diagnóstico da insuficiência
suprarrenal. O ACTH é usado no tratamento da esclerose múltipla e
do espasmo infantil (síndromeWest).
3. Efeitos adversos. A toxicidade é similar à dos glicocorticoides e in
cluem osteoporose, hipertensão, edema periférico, hipopotassemia,
distúrbios emocionais e aumento do risco de infecções.
B. Hormônio do crescimento (somatotropina)
A somatotropina é um grande polipeptídeo que é liberado pela hipófise
anterior em resposta ao hormônio liberador do hormônio do crescimento
produzido no hipotálamo (verFigura23.2).A secreção do hormôniodo cres
cimento (GH, de growth hormone) é inibida por outro hormônio hipofisário,
a somatostatina (ver a seguir). O GH é liberado de modo pulsátil, com os
níveis mais elevados ocorrendo durante o sono. Com a idade, a secreção
de GH diminui, sendo acompanhada de uma redução na massa muscular
magra. A somatotropina influencia uma ampla variedade de processos bio
químicos; por exemplo, pela estimulação da síntese proteica são promovi
dos a proliferação celular e o crescimento ósseo. O aumento da produção
de hidroxiprolina a partir da prolina reforça a síntese de cartilagem. O GH
humano sintético é produzido portecnologia de DNA recombinante e deno
minado somatropi
na. O GH de fontes animais é ineficaz no homem.
1 . Mecanismo de ação. Embora vários efeitos fisiológicos do GH se
jam exercidos diretamente nos seus alvos, outros são mediados
pelas somatomedinas - fatores de crescimento tipo insulina 1 e li
(IGF-1 e IGF-11, de insuli
n-like growth factor). (Nota: na acromegalia
[uma síndrome do excesso de hormônio do crescimento], os níveis
de IGF-1 são consistentemente elevados, refletindo GH elevado.)
2. Usos terapêuticos. A somatotropina é usada no tratamento da de
ficiência de GH no crescimento em crianças. É importante estabele-
,
,
Farmacologia Ilustrada 293
,
HIPOTÁLAMO
LOBO
POSTERIOR.--
CRH
HIPÓFISE
,
SUPRARRENAL
,
,
,
,
,
,
· - .
• •
•
•
•
•
LOBO
ANTERIOR
Pró-oplo
melano
cortina
ºt
ACTH
· - .
• • •
........
Colesterol -
-
º
-
-
)
• Pregnenolona
Desmolase i
SUPRARRENAL
Ações
anti-inflamatórias
Aumentaa
gliconeogênese
Aumenta
ahidrólise
de proteínas
Progesterona
�
t
Cortisol
Cortisol
Figura 23.3
Secreção e ações do hormônio adreno
corticotrópico (ACTH). CRH = hormô
nio liberador de corticotropina.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-303-320.jpg)





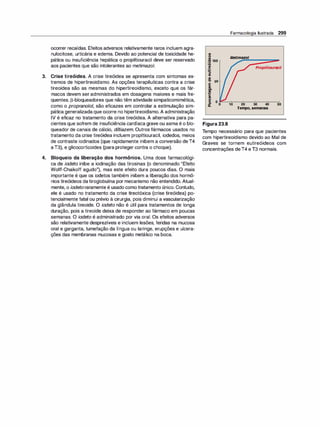


![302 Clark, Finkel, Rey & Whalen
Tipo 1 Tipo 2
Idadeno Geralmente Frequentemente
início duranteainfância acimados
oua puberdade. 35anos.
Estado Frequentemente Geralmente
nutricional subnutrido. presençade
noinício obesidade.
Prevalência 5a 10%dos 90a95°/odos
diabéticos diabéticos
diagnosticados. diagnosticados.
Predisposição Moderada. Muitoforte.
genética
Defeitoou Ascélulas13são Incapacidadedas
deficiência destruídas, células13de
eliminandoa produzir
produçãode quantidades
insulina. apropriadasde
insulina; resistência
àinsulina;outros
defeitos.
Figura 24.2
Comparação entre os diabetes tipo 1 e
tipo 2.
Indivíduosnormais
m
e
·
-
::::1
Ul
e
·
-
-8
rJ
·-
40
;;
E
!(!
i5..
o
'ti
m
...
/
Diabetestipo2
Diabetestipo1
li o ��---�----�
u
� o 5 10
o Minutos
I
--
-
-
r:r
Infusãode glicose
Figura 24.3
1
Liberação de insulina que ocorre em
resposta a uma carga de glicose por
via IV em indivíduos normais e em pa
cientes diabéticos.
dependente de insulina), diabetes gestacional e diabetes devida a outras cau
sas (p. ex., defeito genético ou medicações)
2
• A Figura 24.2 resume as carac
terísticas do diabetes tipo 1 e tipo 2. O diabetes gestacional é definido como
intolerância aos carboidratos com início ou primeiro reconhecimento durante a
,
gestação. E importante manter controle glicêmico adequado durante a gesta-
ção, pois o diabetes gestacional descontrolado pode causar macrossomiafetal
(corpo anormalmente grande), distocia de ombro (dificuldade no parto), bem
como hipoglicemia neonatal. Dieta, exercício e/ou administração de insulina
são eficazes nessa condição. Gliburida e metformina podem ser alternativas
razoavelmente seguras ao tratamento com insulina para o diabetes gestacio
nal. Contudo, grandes estudos randomizados são necessários para avaliarde
finitivamente o resultado neonatal e o regime de dosagem ideal.
A. Diabetes tipo 1
O diabetes tipo 1 atinge mais comumente indivíduos adultos jovens ou
que estão na puberdade, mas algumas formas latentes podem ocorrer
mais tardiamente. A doença se caracteriza por uma falta absoluta de in
sulina causada por extensa necrose das células J3. A perda da função
das células J3 em geral é atribuída a processos autoimunes direcionados
contra essas células e pode ser deflagrada por invasão virai ou ação de
toxinas. Como resultado da destruição dessas células, o pâncreas deixa
de responder à glicose, e o diabetes tipo 1 mostra os sintomas clássicos
da deficiência de insuli
na (polidipsia, polifagia, poliúria e perda de massa
corporal). O diabetes tipo 1 exige insuli
na exógena para evitar o estado
catabólico que resulta e é caracterizado pela hiperglicemiae a cetoacido
se ameaçadora à sobrevida.
1. Causas do diabetes tipo 1 . No período pós-absortivo, são man
tidos níveis baixos basais de insulina circulante pela secreção
constante das células J3. Isso suprime a lipólise, a proteólise e a
glicogenólise. Uma onda de secreção de insulina ocorre dentro de
2 minutos da ingestão do alimento, em resposta ao aumento transi
tório dos níveis de glicose e aminoácidos circulantes. Isso dura por
até 15 minutos e é seguido de secreção pós-prandial de insuli
na.
Contudo, não possuindo células J3 funcionais, o diabético tipo 1 nun
ca consegue manter um nível de secreção basal de insulina e nem
responde às variações de energia circulantes (Figura 24.3). O de
senvolvimento e a evolução da neuropatia, nefropatia e retinopatia
estão relacionados diretamente com o grau de controle glicêmico
(medido como glicemia e/ou hemoglobina A1c [HbA,c]).
3
2. Tratamento. A pessoacom diabetes tipo 1 depende da insulina exó
gena (injetada) paracontrolara hiperglicemia, evitara cetoacidose e
manter níveis aceitáveis de hemoglobina glicosilada (HbA1c)· (Nota:
a velocidade de formação de HbA1c é proporcional à média da gli
cemia nos três meses prévios. Por isso a HbA1c serve como medida
de quão bem o tratamento normalizou a glicemia no paciente dia
bético.) O objetivo da administração de insulina ao diabético tipo 1
é manter a concentração de glicose tão próxima do normal quanto
possível e evitar as variações amplas entre seus níveis de glicose
que podem contribuir para as complicações de longo prazo. O uso
de monitores de glicemia domésticos facilita a automonitoração fre-
2
Ver Bioquí
mica ilustrada, 4ª edição, Artmed Editora, para uma discussão
sobre o diabetes tipo 1 e tipo 2.
3Ver Bioquí
mica ilustrada, 4ª edição, Artmed Editora, para uma discussão
sobre hemoglobina A1c.](https://image.slidesharecdn.com/farmacologiailustrada5ediao-220410140527/85/Farmacologia-ilustrada-5-edicao-pdf-312-320.jpg)

















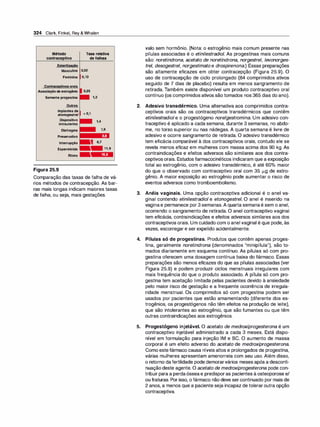









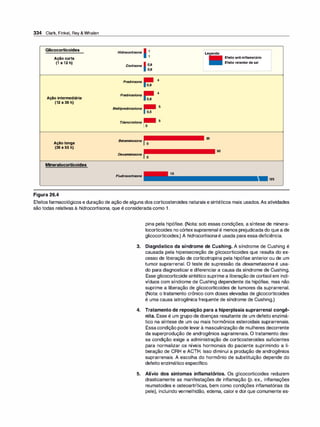

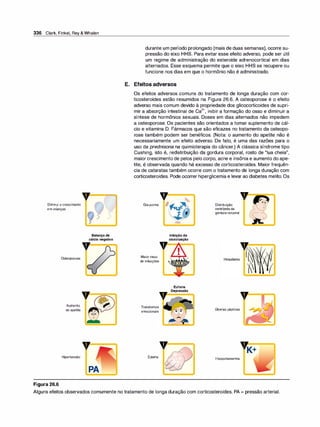

















































































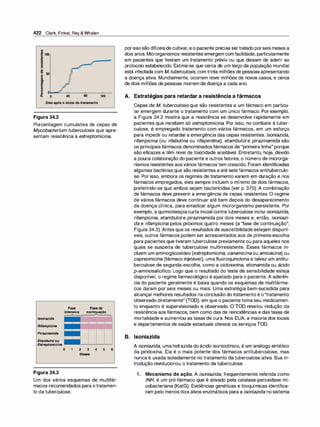













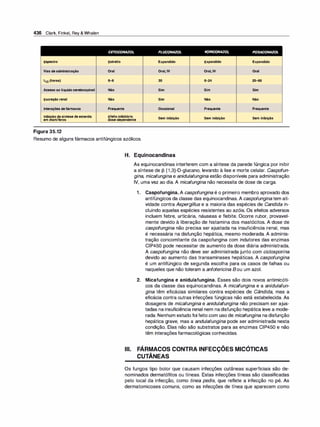
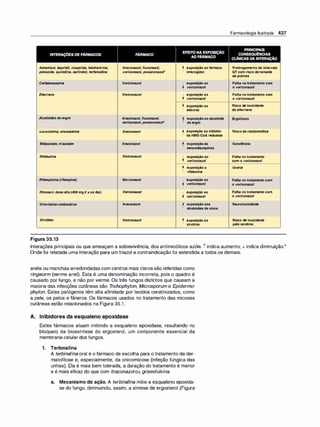




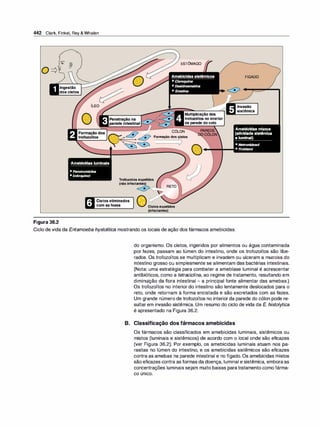














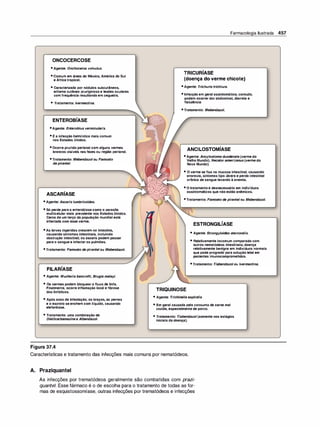











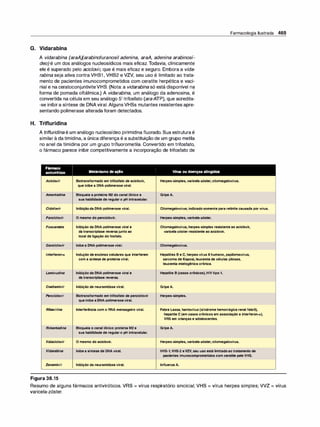














































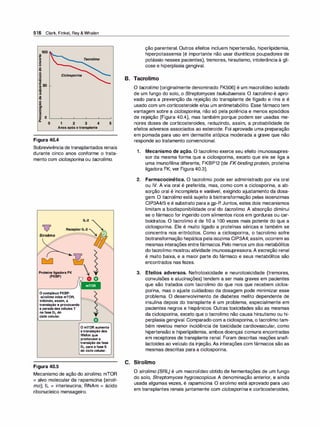






































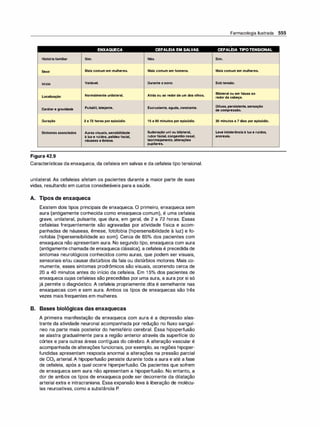

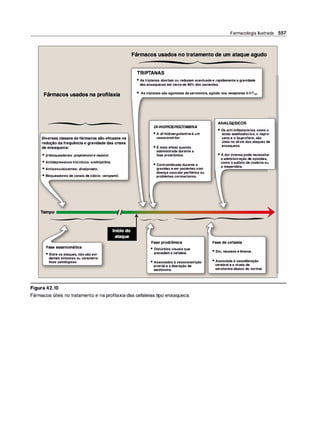
















































































![Questões de farmacologia (dissertativas e objetivas) [pré teste fcms]](https://cdn.slidesharecdn.com/ss_thumbnails/questesdefarmacologiadissertativaseobjetivaspr-testefcms-161211034124-thumbnail.jpg?width=640&height=640&fit=bounds)









































